ArchDB is a classification of loops extracted from known protein structures. The classification of loops in archDB is based in the length of the loop, its conformation (φ and ψ backbone dihedral angles of the residues in the loop), the distance between the extremes of the loop, the bracing secondary structures of the loop and the geometry defined by the super-secondary structure motif (the loop itself plus the bracing secondary structures)
We have defined a loop as a super-secondary structural motif, consisting of an aperiodic structure connecting two sequential periodic secondary structures. In ArchDB, is the basic unit for classification. For the ArchDB classification purposes, a loop is defined by the number of residues forming the aperiodic structure, its conformation (φ and ψ backbone dihedral angles of the residues in the aperiodic structure), the bracing secondary structures of the loop, and the geometry of the loop.
The geometry of the loop is defined by four internal coordinates (D, δ, θ, ρ) extracted from the orientation of the principal vectors (M1, M2) that define the bracing secondary structures (see Oliva et al. 1997):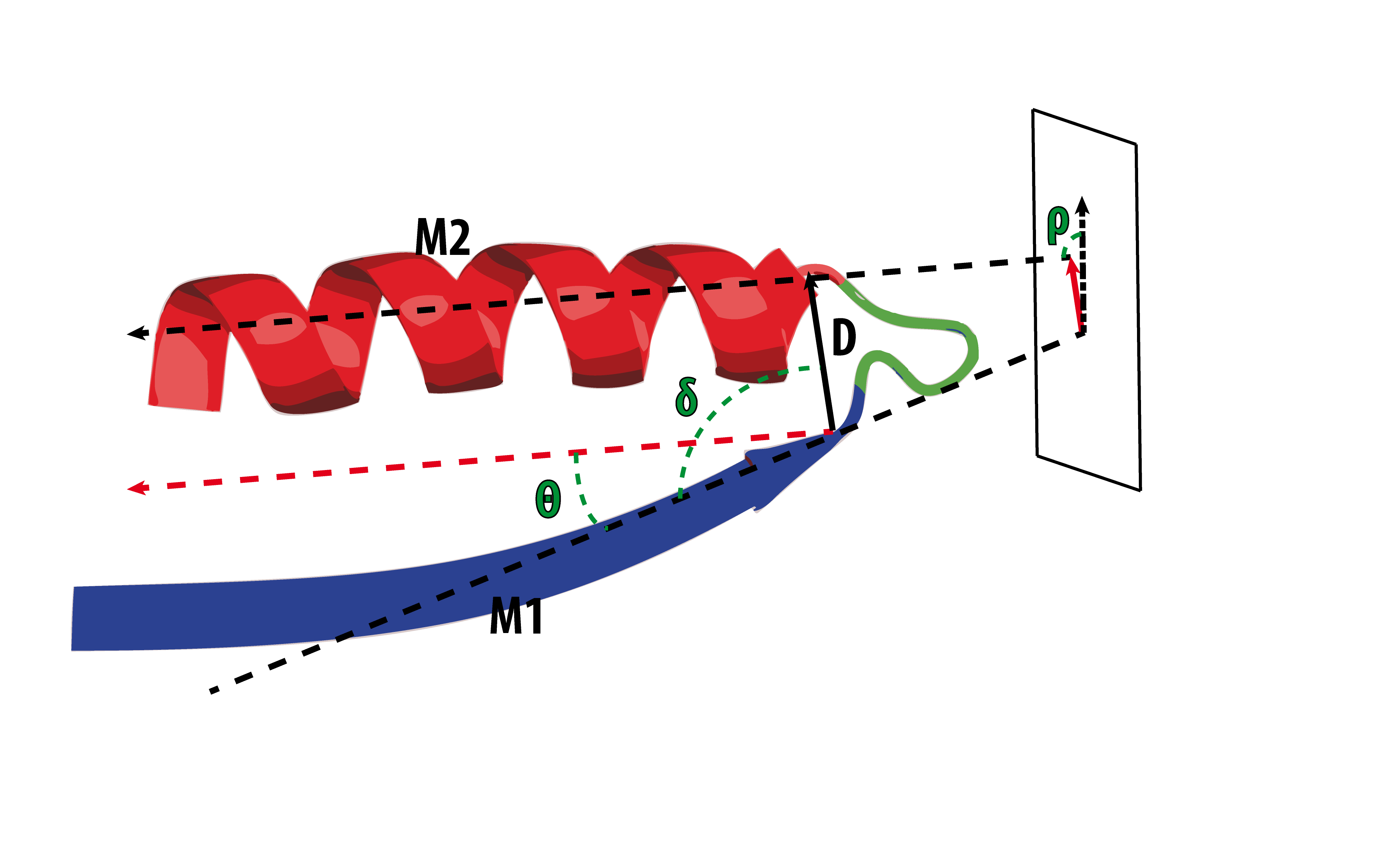
The geometry of the loop is defined by four internal coordinates (D, δ, θ, ρ) extracted from the orientation of the principal vectors (M1, M2) that define the bracing secondary structures (see Oliva et al. 1997):
- D: Distance. The euclidian distance between the boundaries of the aperiodic structure.
- δ: Delta (hoist) angle. The angle between M1 and D.
- θ: Theta (packing) angle. The angle between M1 and M2.
- ρ: Rho (meridian) angle. The angle between M2 and the plane Γ defined by the vector M1 and the normal to the plane formed by M1 and D.
The previous version of ArchDB (Espadaler et al. 2004) only used the Density Search (DS) clustering method, allowing for a potential extension of ± 1 residue in the aperiodic structure of the arch within clusters, representing the potential variability in residue length of geometrically similar loops. Due to the enormous increase of experimental data, implementing such potential extension was timely unfeasible in the current version of the database.
However, the variability feature implemented in the previous version of the database is a requirement if one pretends to identify geometrically similar loops with different residue length. To surmount this problem, in the current version of the database, we implemented a new clustering approach, the Markov CLustering algorithm (MCL) that makes feasible to cluster together loops with different residue length. The DS clustering is maintained for consistency with the previous version of the database, and to enlarge the coverage of clustered loops.
However, the variability feature implemented in the previous version of the database is a requirement if one pretends to identify geometrically similar loops with different residue length. To surmount this problem, in the current version of the database, we implemented a new clustering approach, the Markov CLustering algorithm (MCL) that makes feasible to cluster together loops with different residue length. The DS clustering is maintained for consistency with the previous version of the database, and to enlarge the coverage of clustered loops.
The Density Search clustering method used in ArchDB is based upon the density or mode-seeking technique (searching for regions containing a relatively dense concentration of loops), a version of single-linkage analysis (Everitt, 1974). Basically, the DS algorithm detects regions with a high density of loops in a features space defined by the length, bracing secondary structures, conformation and geometry of the loops. All loops in DS clustering have identical bracing secondary structures and number of amino-acids in the aperiodic structure of the arch, a consensus conformation (nearly identical), and a similar geometry.
The Markov CLustering algorithm (MCL) is a graph-based clustering algorithm. The idea behind the MCL algorithm is to simulate a flow of information within the graph, enhancing the flow where the current is strong and hindering it where the current is weak. If natural groups are present in the graph, then streams across borders between different groups will fade out. In MCL, the flow is controlled expanding and inflating the stochastic (Markov) matrix that represents the graph (see Van Dongen 2008).
In ArchDB such graph is built considering loops as vertices and setting an edge between two loops if their conformation and geometry are similar. An edge between two loops is established when all the following conditions are met:
In ArchDB such graph is built considering loops as vertices and setting an edge between two loops if their conformation and geometry are similar. An edge between two loops is established when all the following conditions are met:
- a minimum percentage of identical phi/psi angles of the loops. This percentage ranges from 95% to 98%
- similar geometrical parameters. Geometrical variation allowed between two linked loops is defined by the four geometric parameters:
- Distance D: ΔD ≤ 1 Å
- Delta angle: Δδ ≤ 15°
- Theta angle: Δθ ≤ 15°
- Rho angle: Δρ ≤ 25°
A class represents different clusters of loops (sub-classes) with identical conformation in the segment defined by the aperiodic structure region plus a minimum of 2 residues of the bracing secondary structures.
If a loop has a 'direct' assignation it means that the loop strcuture (exctracted from the corresponding PDB file) was directly used during the classification. In other words, such loop structure had undergone the classification process by DS and MCL algorithms (but it does not mean that such loop was actually classified).
When two chains of a PDB file have identical sequences (i.e.: A, B) only one of them (i.e.: A) is handled during the clustering process. Then the classification obtained for the loops of the processed chain (i.e.: A) is transferred by ‘identity’ to the corresponding loops of the non-processed chain (i.e.: B).
Classified loops are assigned to redundant PDB sequences through sequence homology by BLAST1. The hits used for the annotation had ot satisfy a minimum percentage of identity according to the length of the alignment (above the twilight-zone curve, as described by Rost2). Only structures with a 100% sequence coverage in the loop region were assigned by this procedure.
1Altschul, S.F., Madden, T.K., Schäffer, A.A., Zhang, J., Zhang, Z, Miller, W & Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res., 25(17), 3380-3402.
2Rost, B. (1999) Twilight zone of protein sequence alignments. Protein Eng., 12(2), 85-94.
2Rost, B. (1999) Twilight zone of protein sequence alignments. Protein Eng., 12(2), 85-94.
For the MCL clustering we initially set five different length groups: small, between 0 and 4 residues; medium, between 4 and 6 residues; large, between 7 and 13 residues, extra-large, between 14 and 20 residues; and extra-extra-large for loops of 21 residues or more. The large population of loops with four residues recommended to cluster them into both the small and the medium group. Hence, a four-residues-length loop may be clustered in sub-classes of the small and the medium groups. This is represented by the notation 4S (in the small groups) and 4M (in the medium groups). Particularly, a 4S sub-class may contain loops within the range of 2-4 residues (including sub-classes constituted exclusively by 4-residues-length loops), while a 4M sub-class may contain loops within the range of 4-6 residues (excluding sub-classes constituted exclusively by 4-residues-length loops, which will be classified in the 4S clusters).
This is because a protein can have more than one of such external DB annotations. In ArchDB, the loop inherits all the annotations from the region of the PDB chain of which the loop is part of. Then, the total number of annotations for a group of loops can exceed the number of loops in such group.
The separated atoms correspond to what PDB understands as HETATM. This are amino acids that do not fit inside the regular 20.
There are 3 different coloring variants for the 3D view (not including the default coloring by secondary structure):
- Amino Acid: This scheme adds one color per Amino acid. Individual aminoacids can be identified by the use of the Labels button
- Shapely: This scheme colors Amino acids according to their traditional properties. The code reads as follows:
Amino Acid(s) Color ASP, GLU bright red CIS, MET yellow LYS, ARG blue SER, THR orange PHE, TYR mid blue ASN, GLN cyan GLY light grey LEU, VAL, ILE green ALA dark grey TRP pink HIS pale blue PRO flesh - Polar: The polar coloring view does not distinguish the sign of the charge, it assigns true to charges and false to non-charged Amino acids.
For some databases it does not make a lot of sense to build a standalone search over ArchDB. For instance, searching by taxid will only reveal the experimental bias over certain species, which would not be very useful when searching for structural properties of proteins.
A loop of length 0 representes two correlative seconday structures (as assigned by DSSP) with no aperiodic structure residues between their boundaries. Thus, it can be found between two secondary structures of different type, like a shift from H (alpha-helix) to G (helix-3:10) or a transition between a beta-strand and an alpha-helix.
The sequence consensus string uses PROSITE format, where positions in sequence are delimited by a dash (-) and the different accepted possibilities for a given sequence position are shown between squared brackets; wildcard positions are represented by an X.
Clustering of loop subclasses is based on the Ramanchandran phi/psi angles and loop geometry. A 9x9 matrix of phi/psi angle is defined as shown in the following table. The codes are explained in the table legend.
Several special symbols can be used in the Ramachandran consensus string:
| b | b | b | p | o | M | e | e | e | 180 | psi |
| b | b | b | p | o | M | M | e | e | 135 | |
| b | b | b | p | * | l | l | s | e | 90 | |
| a | a | a | T | * | l | l | g | N | 45 | |
| N | a | a | a | * | U | l | g | N | 0 | |
| N | a | a | a | * | U | g | g | N | -45 | |
| I | a | a | a | * | G | G | G | I | -90 | |
| e | F | F | F | o | e | e | e | e | -135 | |
| b | b | b | p | o | e | e | e | e | -180 | |
| -180 | -135 | -90 | -45 | 0 | 45 | 90 | 135 | 180 | ||
| phi | ||||||||||
* : forbidden regiona : alpha helixl : left handed alpha helixb : beta strandp : beta prolineg : gammae : epsilonl/g : bridge region between left handed and gamma helices
(written as'v' in loops multiple alignment)b/p : bridge region between beta strand and beta proline
(written as'x' in loops multiple alignment)
- dash (-): represents a cys-proline.
- dot (.): represents a position for which no consensus was achieved.
The assignation of external codes to loops varies depending on the source database:
taxid and enzyme: These two relations aredirectly assigned to thefull chain . This means that the relation is extracted directly from the PDB source and assigned to all the loops of that specific chain.uniprot and SCOP: These relations aredirectly assigned positionally . This means that the relation between a PDB chain and those codes is assigned through direct reference by any of the mentioned databases but only to the specified position of the chain. Only loops fully inside the assigned region are assigned that specific reference (both loop limits belong to the assignation).GO and Drugbank: These relations areassigned through uniprot . In those two cases, it is the uniprot assignation the one that links a PDB to those databases. Thus, the assignation is dependent of the same conditions as the uniprot assignation.
ChemDoodle is a JavaScript library that requires the browser to support a number of components. For most of them, a simple update of your browser will suffice to correctly use ChemDoodle. However, one of such components, WebGL, may require further installation. Here, we provide a few hints on how to enable WebGL in some popular browsers. For further advice, please refer to the distribution site to get installation instructions specific for your browser.
A quick overview on how to activate WebGL in the last versions of some popular browsers:
Safariopera:gpu ' in your browsing bar and check your WebGL status. If any error or warning is reported, we suggest you to use any other of the referred browsers.
A quick overview on how to activate WebGL in the last versions of some popular browsers:
Safari
- Click on the settings icon and select Preferences.
- Click the Advanced tab in the Preferences window.
- At the bottom of the window, check the Show Develop menu in menu bar checkbox.
- Open the Develop menu in the menu bar and select Enable WebGL.
- Type chrome://flags where you would normally type a web address. Hit enter
- Under the Experiments list, find "Override software rendering list" and press Enable.
- Type about:config into the browser and press Enter where you would normally type a web address.
- Type webgl.force-enabled into the Filter: search box.
- Press your mouse on the Value that is displayed to highlight the row.
- Right click your mouse on the highlighted line and choose Toggle.
- Click "Options" from the menu or write '
settings ' in the browsing bar. - Enable hardware acceleration.
- Enable WebGL (use the search tool if necessary; if you cannot find it, please read the note at the bottom of this section)
- Restart your Opera browser