Help Page
Introduction:
Conformational clusters and consensus sequences have been derived
by computational analysis for loops from
SCOP 40 database.
Loops have been classified into five types (alpha-alpha, beta-beta links, beta-beta hairpins,
alpha-beta and beta-alpha) according to the secondary structures they
embrace.
Geometry
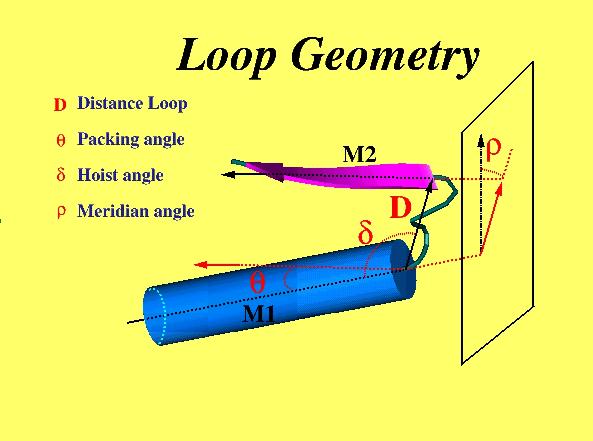
Four parameters were defined to describe the protein loop geometry.
An axis for an alpha or beta secondary structure was defined based on the
shortest of the principal moments of inertia of that structure. The
geometry of each motif was defined by four internal co-ordinates. M1 and M2
are the axis vectors of the secondary structure. Loop geometry is defined
by four parameters (The angles are in greek font in the figure).
- D - distance between alpha helix or beta strand which brace the
loop.
- Theta - the packing angle, i.e, the angle between axis M1 and M2.
- Delta - the hoist angle, i.e., the angle between axis M1 and
vector of D.
- Rho - the meridian angle, i.e., the angle between axis M2 and the plane.
Five tables of loop classes were constructed based on the types of
loop. The table headers are explained as follows:
- Class: -The class of loops defined by the loop clustering in the
paper, e.g., aa{b}aa is a loop that links two alpha helices with
residues of beta Ramachandran angle in the loop.
- CN: -Class number, a numerical number for the definition of the class,
e.g., 1.1.1 and so on.
- NR: -Number of residues in the loop. This number may vary in the same
loop class.
- RMSD(Å): -Root Mean Square Deviation of the superposed loops and
their bracing secondary structure. It is used to quantify agreement
but not for clustering. To calculate the RMSD all residues included
in the multiple sequences alignment are taken into account
(see "Alignment length").
- Consensus: -Sequence Consensus within the loop class. Capital
letters for conserved residues, such as G - Glycine, D - Aspatic
acid. p for polar residues [D, E, H, N, Q, R, S, T, Y]; h for
non-polar residues [A, C, F, G, I, L, M, P, V, W, Y]. Tyr (Y) is
assigned for both polar and non-polar residues. X denotes no
sequence consensus.
Ramanchandran Angles:
Clustering of loop subclasses is based on the Ramanchandran phi/psi
angles and loop geometry. A 9x9 matrix of phi/psi angle is defined as
shown in the following table. The codes are explained in the table legend.
| b |
b |
b |
p |
o |
M |
e |
e |
e |
| b |
b |
b |
p |
o |
M |
M |
e |
e |
| b |
b |
b |
p |
. |
l |
l |
s |
e |
| a |
a |
a |
T |
. |
l |
l |
g |
N |
| N |
a |
a |
a |
. |
U |
l |
g |
N |
| N |
a |
a |
a |
. |
U |
g |
g |
N |
| I |
a |
a |
a |
. |
G |
G |
G |
I |
| e |
F |
F |
F |
o |
e |
e |
e |
e |
| b |
b |
b |
p |
o |
e |
e |
e |
e |
|
|
|
|
|
- a: alpha helix
- l: left handed alpha helix
- b: beta strand
- p: beta prolin
- g: gamma
- e: epsilon
- l/g: bridge region between left handed and gamma helices
(written as 'v' in loops multiple alignment)
- b/p: bridge region between beta strand and beta prolin
(written as 'x'in loops multiple alignment)
|
Pattern:
Weighted aminoacid frequencies (according to Henikoff & Henikoff, 1994) were calculated for each position. Then, aminoacids were grouped into predefined sets.
If the sum of individual frequencies for a given set was equal o greater than 75%
and lower than 90%, then such aminoacids are
shown between brakets. If the sum of individual frequencies was equal or
greater than 90%, then shuch aminoacids are shown between square brakets.
Note that only the aminoacids present at that given position are shown, and not the
full set.
Bracing secondary structures were included in motif calculation.
Conservation degree:
Position specific conservation degree was calculated with
AL2CO
(Pei J. & Grishin N.,
Bioinformatics 2001).
The options used were independent-count method for frequencies calculation and sum-of-pairs method
(using a BLOSUM62 matrix)
for conservation calculation. Bracing secondary structures were included in conservation calculation.
Specifics for ArchDB-KI
Functional Residues
Functional annotation of residues was classified in four categories:
- ATP interaction: for residues involved on ATP binding/interaction.
- Substrate binding: for residues involved in substrate interaction/binding with the exception of ATP
- Ion interaction: for residues involved in ion interaction/binding of ions needed for the catalytic mechanism
- Catalytic: involved in reaction, the stabilization of a transition state or the activation of substrates.
Three different approaches were applied to identify functional residues of the loops of the sub-classes:
- Residues found within a cut-off distance of 6Å from an heteroatom, ligand, inhibitor, cofactor or complex partner molecule (protein or DNA), with the exception of D2O or crystallization buffer molecules.
- Residues identified by functional information from ACTSITE and SITE records in the RCSB protein data bank.
- Residues identified by the functional annotation collected from the literature and assigned to specific motifs of kinases.
Functional Subclasses
We considered loop subclasses as functional subclasses, where there is a meaningful conservation of functional residues in the loops of the cluster, and more than 50% of loops belong to the same SCOP superfamily.
Browsing and Querying ArchDB
- Search by sequence: Users can search for classified loop(s) with sequence similarity to a query sequence.
- Search by structure: Users can upload protein coordinates in PDB format and its loops will be extracted and compared with those from the classification. First: structural class is assigned comparing loop geometry and, second: the loop conformation is compared among the subclasses within the assigned class.
- Search subclasses and/or Search Loops: A range of options are offered for subclass or loop searches. Users can query ArchDB asking for subclasses or loops with specific flanking secondary structures, length of loops or phi/psi loop conformation. Also, users can retrieve all subclasses or loops with PDB SITE annotations and contacts with co-crystallized ligands. Finally, users can search for subclasses that have SCOP, GO or EC annotations conserved at different percentage levels.
- Search structures: Users can search for classified pdb structures in ArchDB with specific Sprot. Annotation/Keyword, GO annotation, SCOP and EC codes.
- Specific queries for ArchDB-KI: Users can list functional subclasses or loops classified in ArchKI.
Pictures of all loop subclasses were generated by superimposing
four loops randomly choosen in each class.
When subclasses contained less than four loops, all loops were included in the picture.
RASMOL Mouse Buttons
- Left - Rotate (X and Y)
- Right - Move
- Shift+Left - Zoom
- Shift+Right - Rotate (Z)
- Up: Zoom Out (smaller)
- Down: Zoom In (bigger)
Instructions for Rasmol script usage
Configuring your browser for the RasMol option (UNIX)
Jmol Interactive Picture:
This a Java applet for easily viewing molecules inside the windows of the browser.
A Java Virtual Machine must be installed in your system. You can
download JRE.
Instructions for Jmol script usage.




