STRUCTURAL STUDY ON ACETOBACTER ACETI CITRATE SYNTHASE
By Clara Tourińo, Eulŕlia Belloc & Alba Duch
INDEX
INTRODUCTION
MODELLING ACETOBACTER ACETI CITRATE SYNTHASE FROM VERTEBRATE TEMPLATES
CONCLUSIONS
REFERENCES
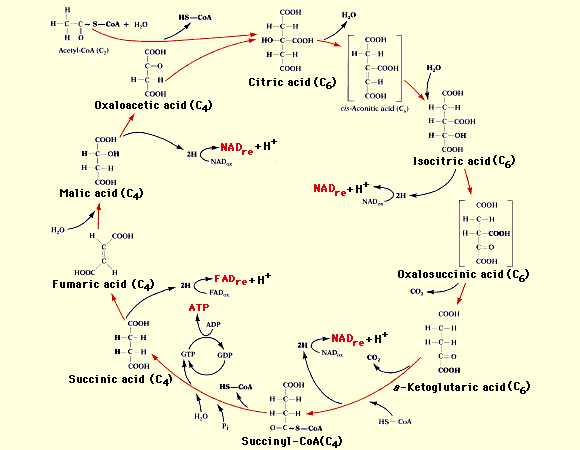
Oxidative respiration is a process which occurs in all aerobic organisms. It is due to this process that aerobic organisms can obtain enough enery to carry out other vital processes. It consists in a set of chemical reactions in which enzimes are really conserved. This fact shows the importance of the pathway. The final step in this path is the Krebs cycle or tricarboxylic acid cycle. In eukariotes, the TCA cycle takes place in the mitocondrial matrix while in prokariotes it takes place in the cytosol because of they lack of different compartments
Any possible substrate for oxidation can be previously transformed into acetil-CoA. With acetyl-CoA the the Krebs cycle can start. Acetyl-CoA binds to an oxalacetate molecule which finally results in a molecule with four carbons: citrate. This first step in the Krebs cycle is carried out by an enzyme named citrate synthase, one of the three irreversible enzymes in the TCA cycle, and one of the control check points. This function is not only carried out in higher forms of life, but also by bacteria and even by members of the achaebacteria phyllums. The active site structure is particularly conserved in organisms with important differences (eg. pig versus archaebacteria).
The classification of citrate synthase is the following:
- Class: All alpha proteins
- Fold: Citrate synthase
- Superfamily: Citrate synthase
- Family: Citrate synthase
- Protein: Citrate synthase
Two distinctly different types of citrate synthase exist in nature:
- The most well studied, are Type I enzymes, they are dimers and can be found in eukaryotes, archaea and gram-positive bacteria.
- In contrast, relatively little is known about Type II citrate synthases. Notably, the Type II enzymes are hexameric, exhibit unique allosteric properties involving acetyl-CoA and NADH, and are found only in gram-negative bacteria.
Actually, all the subunits of the protein are identical, which means that all citrate synthases are homodimers or homohexamers. Furthermore, the subunits from dimers and hexamers are equivalent (49 kDa). Each monomer comprises a large domain, containing 15 helices, and a small domain, containing 5 helices, with the active site cleft between them.
The Acetobacter aceti's citrate synthase, named acid acetic resistant, is a Type II enzyme, so is an hexamer.
Chemical reaction details
THE ACTIVE SITE
The active site of citrate synthase consists in three crucial residues: two histidines and one aspartic acid. The surrounding residues also interact in the catalysis but their contribution is not fully understood. Histidine 274, Histidine 320, and Aspartic Acid 375 in the active site act to hold oxaloacetate and acetyl CoA in the proper conformation for catalysis. Each residue has inherent function in the active site.
- His 320 holds the carbonyl of oxaloacetate, the alcohols of Citryl-CoA, and the citrate through a hydrogen bond interaction.
- Asp 375 is in the acetyl-CoA binding domain which brings about the formation of an enolate intermediate.
- His 274 makes two different bonds, stabilizing both the thioester carbonyl of acetyl-CoA and the carboxylate of citrate after hydrolysis of the thioester intermediate.
The oxaloacetate binding site lies in a cleft formed by the two domains of one subunit. After the condensation reaction between acetyl-CoA and oxaloacetate, citrate is produced. Hence this site is also the binding site for citrate. Citrate is bonded to 3 histidine residues from one subunit. These are histidine residues 274, 238, 320.
The Coenzyme A binding site, like the oxaloacetate binding, involves both subunits. Enzymes used in the bonding of Coenzyme A are His 238, His 274 and His 320 Arg 329, Arg 401.
After the initial binding of oxaloacetate to the enzyme active site, citrate synthase undergoes a conformational change smaller domain seems to rotate 18° moving over the larger one. Then, residue His 320 is believed to cause the polarization of the oxalacetate carbonyl bond, making it more acceptable for interactions with acetyl-CoA. Next, the binding of Acetyl-CoA to the citrate synthase-oxalacetate complex precedes the acid-base reaction between His 274 and Asp 375. Further hydrolyzation of citryl-CoA yields citrate and CoA.
As said before, citrate synthase is an enzime from a very conserved pathway, the Krebs cycle. This pathway is present in all the aerobic organisms, with the same role, transforming oxalacetate and CoA into citrate. The aim of the study is to prove whether a citrate synthase from an extremophile organism has the same structure, the same function and a similar sequence that one from such a different organism as a vertebrate or not. The project will focus in studying the sequencialchanges between different species enzyme and specially the conformational changes in the active site during all the catalytic process. To do this, a sequence from Acetobacter aceti citrate synthase will be modelled using templates from Gallus gallus (chicken) and Sus srofa (pig). Furthermore, a reproduction of the conformational changes of the ezyme, specially in the active site will be reproduced in the A. aceti protein.
This study is based on the modelling of the problem sequence citrate synthase acid acetic resistant whose Swissprot code is P20901.
To achieve the model we followed two strategies:
Strategy A.
- Query submission in Blast against Swissprot database, and choice of sequences below an E-value treshold of e-04.
- Alignment of the previous sequence with Clustalw.
- Construction of a Hidden Markoff Model using the Clustalw alignment using the software HMM build.
- Search in PDB database using the Hidden Markoff Model and the HMM search software. The sequences obtained are the following:
- 1cts
- 4cts chain A
- 2cts
- 6cts
- 6csc chain A
- 1al6
- 1amz
- 1csh
- 1csi
- 1csr
- 1css
- 6csc chain B
- 1csc
- 2csc
- 3csc
- 4csc
- 5cts
- 6cts
- 5csc
- 1aj8 chain A
- 1aj8 chain B
- 1a59
Strategy B.
- Query submission in Blast against PDB and choice of sequences below an E-value treshold of e-04.The sequences obtained are the following:
- 1cts
- 4cts chain A
- 4cts chain B
- 1al6
- 1amz
- 1csr
- 1css
- 6csc chainB
- 1aj8 chain A
- 1csc
- Selection and clustering of the template sequences depending on which heteroatoms they are complexed with looking at the information in SCOP.
As the aim of the study was to picture out the sequence of the enzymatic reaction, the sequence choice from blast results was following the next criteria: On the one hand complexed with the substrate, product, and coenzyme, and on the other the non complexed enzyme. Due to the most suitable structures were present in both search strategies, the study continued with a unique strategy using these sequences:
- Open conformation: template 5cscA.
- Complex with oxalacetate and acetyl-CoA conformation: template 1al6 and 5cts.
- To model quence in a complex with citrate and acetyl-CoA conformation: template 2cts and 6cscA.
- Complex with citrate conformation: template 1cts.
This coincidence reflects the strength of the choice. But it is important to note that the complexed molecules were not the real substrates and coenzymes but were inhibitors, or analogs, because the cristalized structure with the proper molecules can't be obtained due to the fact that the reaction would progress normaly, so that a relatively static complexed molecule is required for cristalization.
The following steps after the sequence choice were:
- Structural alignment with the STAMP software with previous sequences.
- Construction of a new Hidden Markoff Model using the STAMP alignment and the software HMM build.
- Alignment of the template sequences and the problem sequence using the previous Hidden Markoff Model and the software HMM align.
- Structural model of the protein P20901 (citrate synthase acetic acid resistant) using MODELLER software (4 models per conformation)
- The energies of the models were revised with Prosa software obtaining a plots which show the energy of each model and within these, each residue. Using these plots, one between the four models is chosen in each conformation, following the criteria of the one with less energy.
- In order to visualize in Rasmol software, the model with the CoA, oxalacetate or citrate the following strategy was used:
- The heteroatoms hold by the template were renamed (HETATM changed to ATOM).
- The template and the model were alignedby means of STAMP software.
- The template was removed from the file obtained in STAMP
- In order to compare structural differences in each catalytic step, an structural alignment was performed again with STAMP software. Because of the fact that some lateral chains were bad modelled, an optimization in the bad modelled lateral chains was performed with GROMOS. Eventually, when all necessary information was collected, they were analyzed and conclusions were reached.
- The models were visualized with the Rasmol program.
Initially, after using the search tools, the pool of selected sequences were both from vertebrates and bacteria, but the latter ones were dropped out from the pool after comparison of sequences in the alignment because of the high diference between the templates. If very different templates were used for modelling a protein, the final model wouldn't have a strong structure. With some different structure in one region, which one would the program would choose? However, after knowing in more detail the stucture and allosteric regulation of our protein, Acetobacter aceti's citrate synthase, a doubt emerged. Choosing bacterial templates instead of vertebrate ones, could have resulted in a better model. But using vertebrate templates to model a bacterial sequences would be usefull to show the conservation of some important enzymes during evolution. Eventually, the aim of the study is to focus on the motion of the active site and see if it it really so conserved,. So, choosing these templates, would it be possible to make a good model from a bacterial protein using templates of such a different organism as a vertebrate?
MULTIPLE ALIGNMENT
To make a multiple alignment sequences from Gallus gallus, Sus scrofa, Antartic bacterium,Acetobacter aceti and Pyrococcus furiosus were selected. With these alignment important differences are seen. There is a very high similarity between the sequences from vertebrates, an a high variability in the bacterial ones, this shows that there has been a great evolutionary divergence. However, the active site residues are highly conserved due to evolutionary constraints to keep the function. Are these evolutionary constraints reflected in the structure? To answer this questions the protein has to be modelled.
PROSA
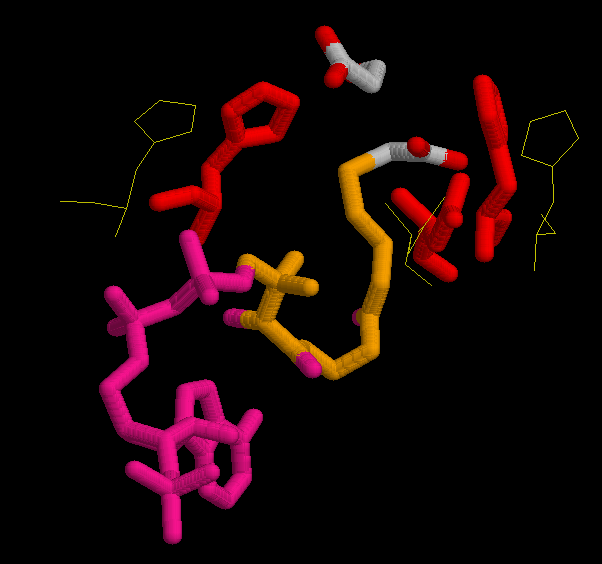
After executing the Modeller program we obtained 4 models for each template. To choose the better one, we analyzed their energies with Prosa software. The pseudopotential energy of the models was in the majority of their residues below zero, which indicates that the models were acceptable. Anyway, there are some regions where the energy is quite high (containing some residues from the active site). At first, it was thought that it was because some basic residues in the active site were linked to negative charges together. I.e. two histidines chained to an oxygen, give rise to very high pseudopotencial energies, because it is very unprobable to find it in an uncomplexed protein. Anyway, these peaks did not appear in the template so this hypothesis was false. Other explanation to these problems had to be found. Eventually, looking at the alignment, this region (arround the resiude 300 from now on) resulted to be the one with more differences in the sequence between the model and the template although when this region was sought in the structure both model and template structure were very similar. In the first picture we can see in red thisregion in the model and in green this region in the template. In the secons picture we can see each residue in a different color. Prosa calculate the energy of the model by means of pseudopotencials looking for the probability that two residues were together. Probably, this region in the Acetobacter aceti's citrate synthase has a different structure, and forcing it to adapt a different conformation making some residues to put together causes a high increase of pseudopotential energies. It would be the possible explanation to this phenomenon.
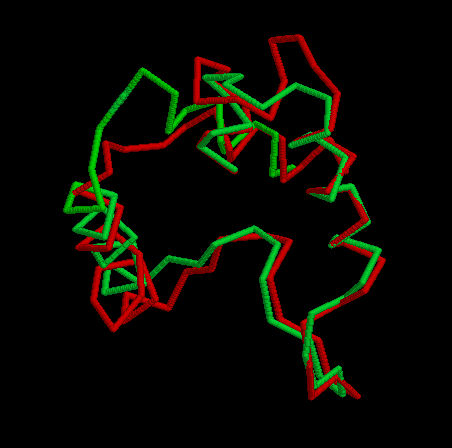

CARACTERIZATION OF THE ACTIVE SITE
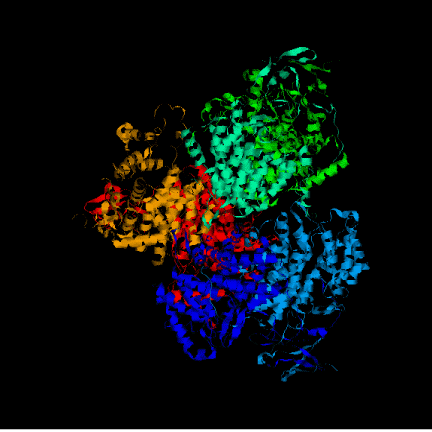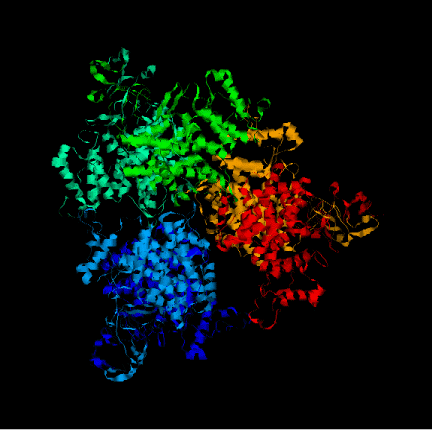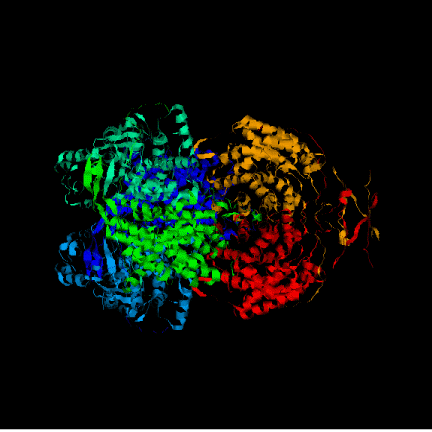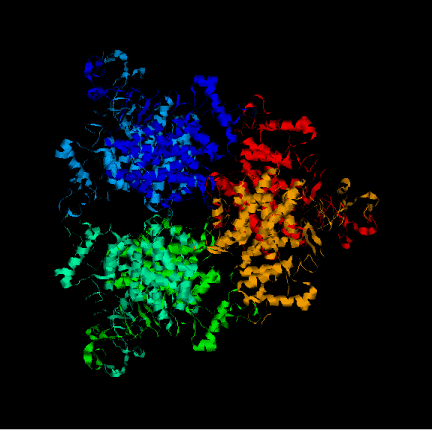
First of all, both conformations of the whole enzyme were compared:
OPEN:________________________________________________________CLOSED:

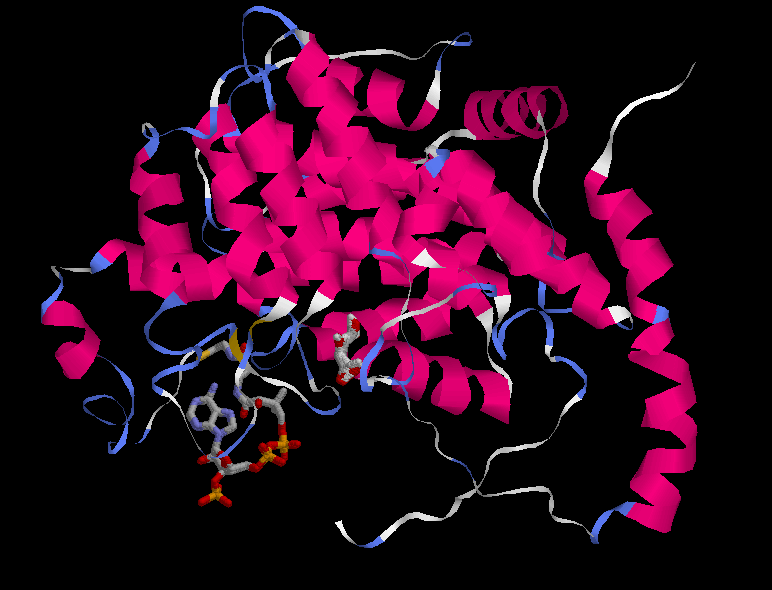
At first sight it is very difficult to see an important conformational change in the protein, although it is known that the citrate synthase changes its structure during the reaction. At the beginning the enzyme remains in an open form, but when oxaloacetate has bound the smaller domain seems to rotate 18° moving over the larger one, closing the cleft and burying the ligand at the active site. The binding of substrate induces a conformational change which generates the acetyl-CoA binding site while simulataneously sealing off solvent access. This is difficult to perceive looking at the whole protein. Instead, the shear motion is much more clear if we focus at particular residues, in concrete the ones in the active site. Anyway, some changes are seen in the pictures of the whole protein. One of the most important one is that of the the beta sheet, what is identified to be part of the hinging mechanism.
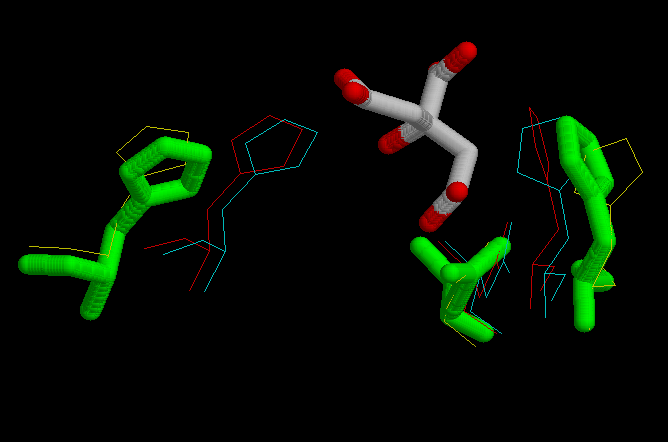
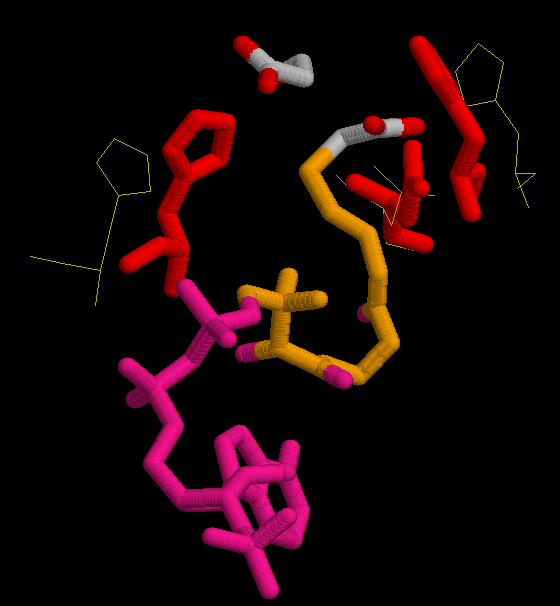
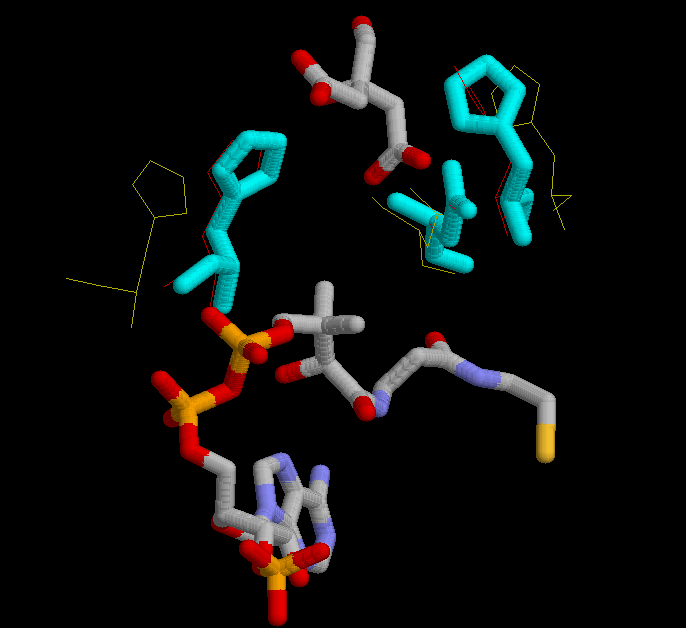
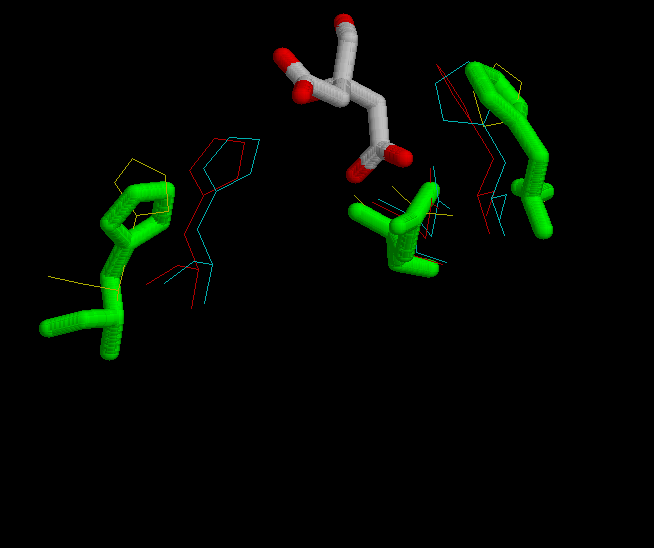
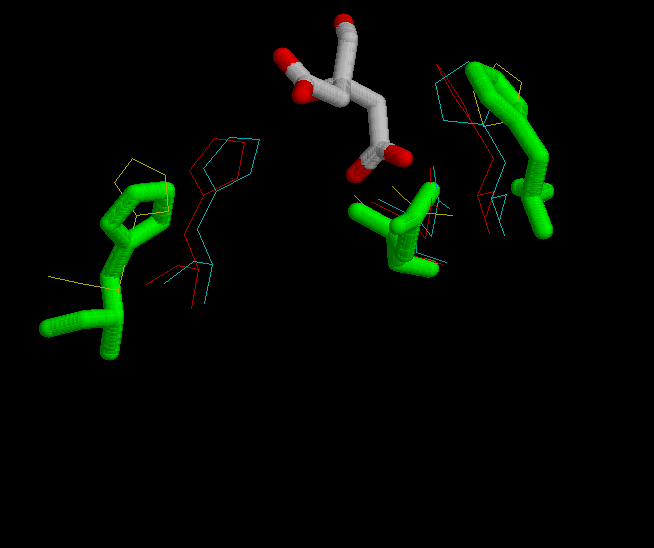
The active sites is located at the interface between the large and small domain of a subunit. There are three crucial residues in the active site responsible from the catalysis. These residues had been mentioned previously, several times. They are His 320, His 274 and Asp 375. So the study will focus on these three residues, and on the changes in conformation that they suffer during de reaction. In the next set of images it it possible to see the motion during the catalytic reaction.
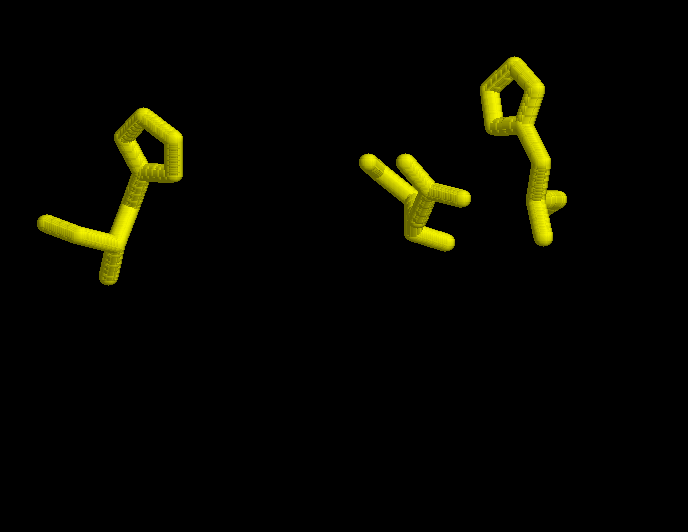
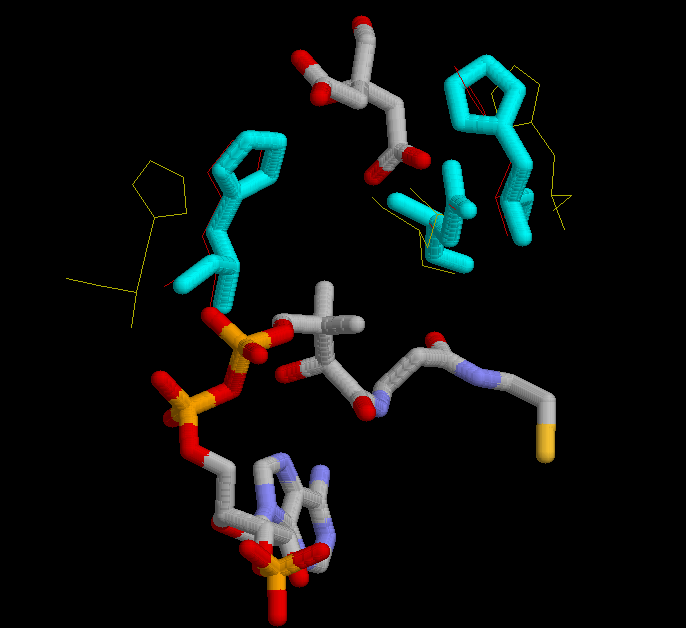
In the first figure, is shown in yellow the possition of His 320, His 274 and Asp 375 in the open form, when neither substrate nor coenzime have bound to the enzyme.
In the second figure, is shown in red the possition of His 320, His 274 and Asp 375 in the complexed form bound with oxalacetate and CoA.
In the third figure, is shown in cyan the possition of His 320, His 274 and Asp 375 in the complexed form bound with citrate and CoA.
In the fourth figure, is shown in green the possition of His 320, His 274 and Asp 375 in the complexed form bound with citrate.
The optimization of the models resulted in another model that didn't fit as well as the ones without optimization. When the optimization was done, some lateral chains adopted strange conformations that were not proper to interact with the target molecule. In the following pictures it is possible to see lateral chains of the optimized model in order to compare it with the non optimized ones above. For instance, let's focus on His 320 in the open model non optimized (above) and optimized (below). The ring has made a rotation in the optimization that would not allow it to interact with its target, the carbonil in oxalacetate. So then, the rest of the work has been done with the non optimized models.
Conformational changes of residues in the active site during the reaction
His 320
When oxaloacetate enters in the cleft that corresponds to the active site, His 320 aproaches to oxalacetate and makes an hidrogen bond with its carbonil. During the reaction it binds to the intermediate citril-CoA staying in the same possition as before, and when the CoA leaves the enzime the histidine keeps holding the group alcohol in citrate but returns to the open possition.
His 274
When subtrate and coenzyme enters, His 274 forms two different bonds, with oxalacetate and with CoA. When the binding takes place the His 274 approaches slightly to the substrate and to CoA (but it moves less than His 320). After hydrolysis of the thioester intermediate, histidine acts stabilizing both the thioester carbonyl of acetyl-CoA and the carboxylate of citrate after hydrolysis of the thioester intermediate, in the same possition. But when the CoA leaves the histidine moves back to the same possition that had in the open form. However, the histidine ring moves independently from de whole chain. It has a rotational movement, depending ont the reaction step. The interaction is maximum when the protein is complexed with citrate and CoA.
Asp 375
The whole chain of Asp 375 does not move during the reaction, but the group COOH does depending on the step.
Other residues in the active site
There are some residues in the active site that in spite of not taking part of the catalytic reaction have an important role in binding the substrate or the CoA. Furthermore, the catalytic residues are also important in the binding of substrate amd coenzime apart from playing a role in catalytic attacks.
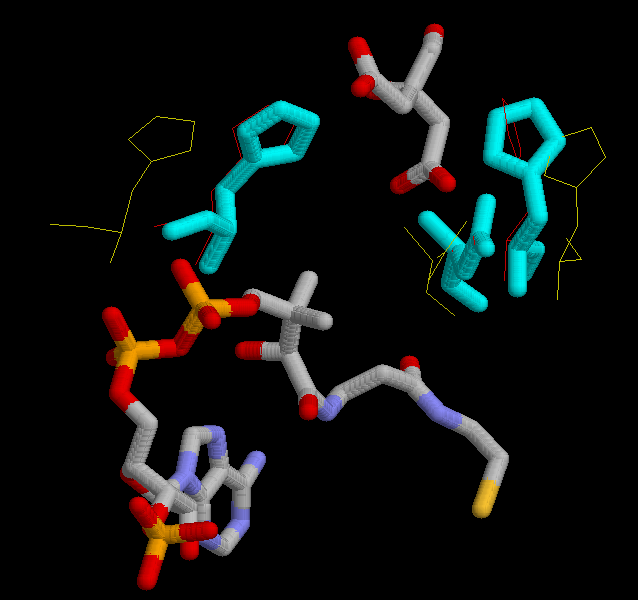
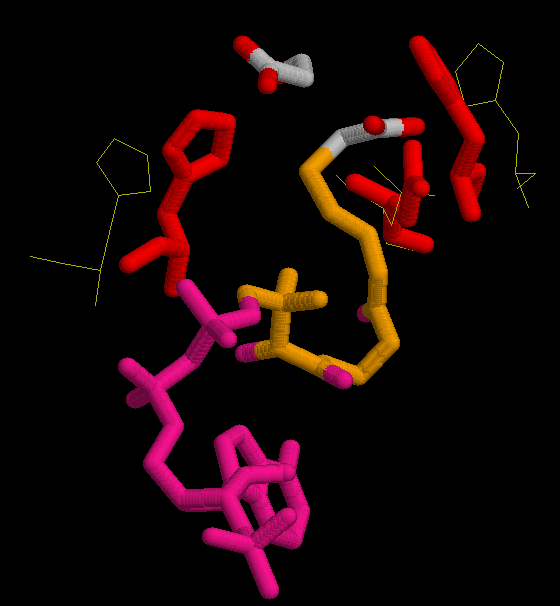
Oxalacetate binds to the protein by the residues His 238, His 274 and His 320.
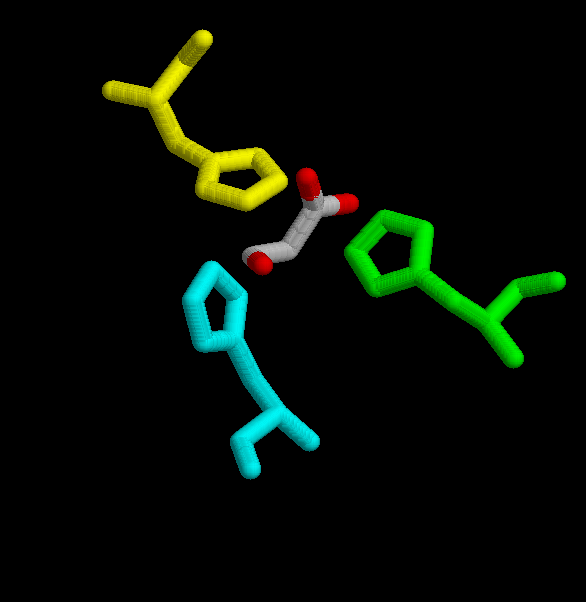
(His 238 -yellow-, His 246 -cyan_, His 320 -green-.)
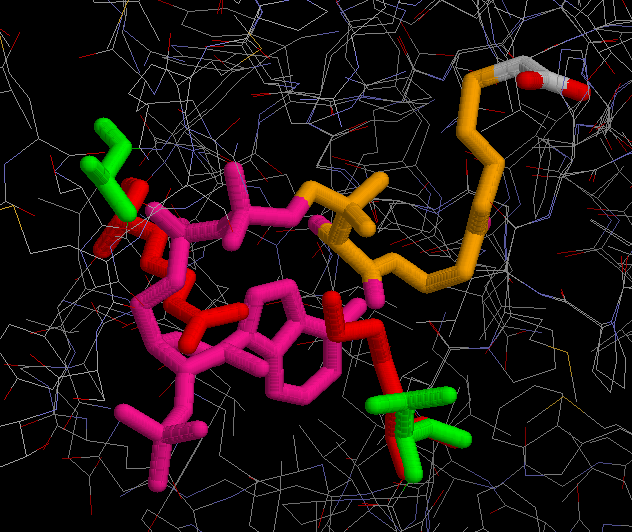
Acetil CoA, is hold by several residues His 238, His 274, His 320, Arg 329, Arg 401. It is also bound to Arg 338 and Lys 281 (in A.aceti).
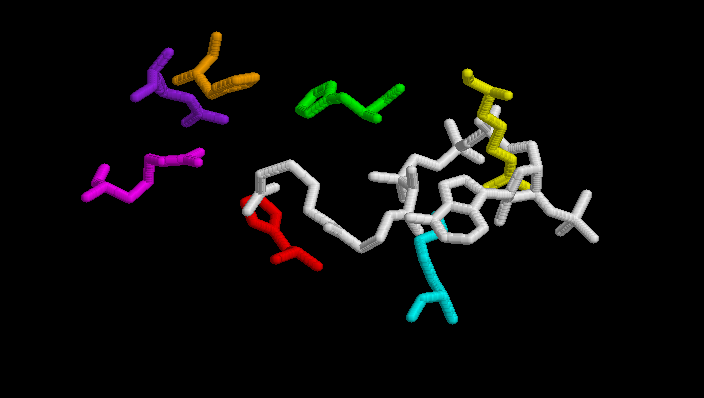
(His 238 -orange-, His 274 -red-, His 320 -green-, Arg 329 -purple-, Arg 401 -magenta-, Arg 338A.aceti -yellow-, Lys 281A.aceti -cyan-)
The next table shows distances between residues in the closed form of the enzyme and substrate or coenzyme. These are possible interactions due to the short distances and the fact that the are charged aminoacids. Some of them have been mentioned before, others not.
| Residue |
substrate or coenzyme |
distance in Amstrongs |
| Asp 375 |
CoA |
2.58 |
| His 274 |
CoA |
3.173 |
| His 320 |
oxalacetate |
4.465 |
| Arg 327 |
oxalacetate |
4.97 |
| Arg 401 |
oxalacetate |
5.45 |
| Gly 275 |
CoA |
4.77 |
| His 238 |
CoA |
2.80 |
| Arg 329 |
oxalacetate |
2.61 |
His 332 |
oxalacetate |
2.89 |
Some of the resiudes that take part in the active site binding pocket remain conserved in the sequence, but some of them have changed, sometimes with afinity enzyme-coenzyme consequences. Oxalacetate molecule binds to the protein by the residues His 211, His 246 and His 287. Acetil-CoA binds to the Arg 296, Arg 370, His 211, His 246 and His 287 in A. aceti protein. Furthermore, Acetil-CoA binds to other residues that have changed in the A. aceti protein. One example is the residue Ala 368 in the chicken protein that has changed to Arg 338 in the A. aceti protein. This residue belongs to the acetil-CoA binding region, so then the A.aceti protein will bind better to the phosphate regions of the acetil-CoA than the chicken one. It is important to note that this arginine is making a bad contact with the Acetil-CoA, what means that its lateral chain has been bad modelled and need optimization. The problem is that we don't have enough tool to make this optimization. Something similar happens with another couple of residues, the Lys 281 of the A. aceti protein and the Val 314 of the chicken protein. This could mean that the binding strength of the A. aceti protein to the acetil CoA is greater than the same enzyme in mammals. (In red residues from A.aceti protein and in green residues from a vertebrate protein).
In spite of the fact that the extremophile A.aceti and vertebrates as pig or chicken are separarated evolutionarily by a great amount of years, they still share some basic mechanisms necessary for using their energy source by means of oxygen. In this study it has been proved that the conservation in one of this mechanisms, the citrate synthase, is so strong that it has been possible to model an enzyme from A.aceti by means of vertebrate templates. Not only the secondary and terciary structure but also lateral chains keep the similar conformation (except some changes in residues). Even it has been possible to reproduce all the conformational changes during the catalytic reaction with enough likelyhood. This may show that bacteria and vertebrates share more traits than may be expected by their morphology, but actually vertebrates and bacteria have common origins, haven't we?
- http://www.idealibrary.com/links/doi/10.1006/jmbi.2001.4808/full
- http://www.chem.uwec.edu/Webpapers2000/Pages/Webpapers2000/roloffam/pages/Mechanism_buttons.html
- Kurz LC, Drysdale GR, Riley MC, Evans CT, Srere PA.Catalytic strategy of citrate synthase: effects of amino acid changes in the acetyl-CoA binding site on transition-state analog inhibitor complexes.
- Kurz, L. C., Shah, S., Frieden, C., et al. Catalytic strategy of citrate synthase: subunit interactions revealed as a consequence of a single amino acid change in the oxaloacetate binding site. Biochemistry 34:13278-13288 (1995).
- Sideraki, V., Wilson, D. K., Kurz, L. C., Quiocho, F. A., and Rodolph, F. B. Site-directed mutagenesis of histidine 238 in mouse adenosine deaminase: substitution ofhistidine 238 does not impede hydroxylate formation. Biochemistry 35:15019-15208 (1996).
- Lasko D, Schwerden C, Bailey J, Sauer U. Acetate-specific stress response in acetate-resistant bacteria: an analysis of protein patterns. Biotechnol. Prog.13:519-523(1997)