TALLER
DE JARDINERÍA.
TALLER
DE JARDINERÍA.
Todo esto está muy bien, pero las colas en los extremos N y C-terminal
nos mosquean un poco. Es claro que el problema está en la secuencia
1emsA,
que es casi el doble de larga. Ante esta crisis estructural nos planteamos
dos posibilidades:
A) Prescindir de este template, aunque tuviera un e-value muy bueno (modelo con un template de menos o mod-1).
B) Recortar del fasta de 1emsA el fragmento que no se alinean con el resto de templates (modelo con el template cortado o modcut).
Del fichero con los fastas de los templates del primer modelo eliminamos
la secuencia 1emsA y grabamos este nuevo fichero con el nombre de temp-1.fa.
De
nuevo alineamos por secuencias mediante CLUSTALW obteniendo un nuevo alineamiento,
mucho más mono como se puede ver, al que llamaremos temp-1.aln.
De igual manera, STAMP alineará los pdb de todos los templates menos
de éste, para obtener HIT-1.pdb.
El programa nos da otra nueva imagen visualizable por RASMOL.
Bueeeeno, pues por lo visto nuestro experimento ha funcionado: como se ve en la imagen, le hemos dado la baja al amiguete 1emsA sin alterar el plegamiento típico de la super-família de proteínas HIT: las 2 hélices y las 5 láminas dispuestas en forma de .............. Procedamos, pues a "fabricar" el modelo:
El HMMER creará la matriz oculta de Markov según estructura para crear temp-1.aliy después lo pasaremos a formato pir y crearemos un top para él solito (mod-1.top).
De nuevo con Modeller, nos dará dos imagenes que llamaremos flori-1A.pdb
y flori-1B.pdby
que visualizaremos por Rasmol.
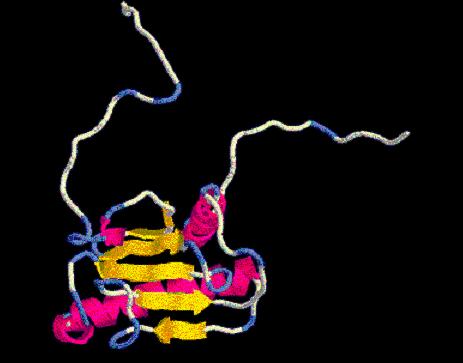
flori-1A.pdb
flori-1B.pdb
¡TENEMOS UN SEGUNDO MODELO!
Antes de ponerse a cortar colas a lo loco habrá que saber por dónde metemos las tijeras.
Para ello, abrimos el alineamiento en CLUSTALW del primer modelo (tempmod1.aln) y vimos que la cola N-terminal llegaba hasta el residuo 268 (fenilalanina), es decir, que hasta el residuo 268 de la cadena 1emsA ésta no tiene homología con el resto del círculo de amigos. Cortamos el pdb por este residuo y lo pasamos por unos script en perl (los programillas PDBtosplitchain y arrageG) que filtran los posibles errores que hayamos podido crear al cortar la secuencia, dándonos los archivos 1emsAcut.fa y 1emsAcut.pdb respectivamente.
Ahora toca repetir todo el proceso. Es decir, repetir el CLUSTALW (obtenemos tempcut.aln), y hacer un nuevo STAMP (obtenemos HITcut.pdb) que visualizaremos por RASMOL.

¿Bonito, eh? Pues aún no ha llegado lo mejor. Ahora con el HMMER, crearemos una nueva matriz oculta de Markov según estructura (modcut.hmm) para alinearla con las secuencias de los amiguetes ( 1emsAestá cortado) y con nuestro protagonista, y obtener tempcut.ali, pasarlo a formato pir y crear un top a su medida (modcut.top).
Con modcut.top
y usando Modeller obtendremos dos nuevas estructuras: floricutA.pdb
y floricutB.pdb.
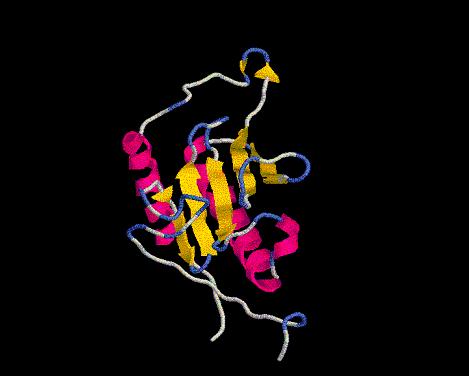
Dicen que a todos tus hijos los quieres por igual. ¿Es ésto cierto con los modelos estructurales? En biología estructural el instinto maternal brilla por su ausencia, así que sólo nos quedaremos con el mejor. Empieza una dura competición...
* PRIMERA PRUEBA (sácame el perfil bueno):
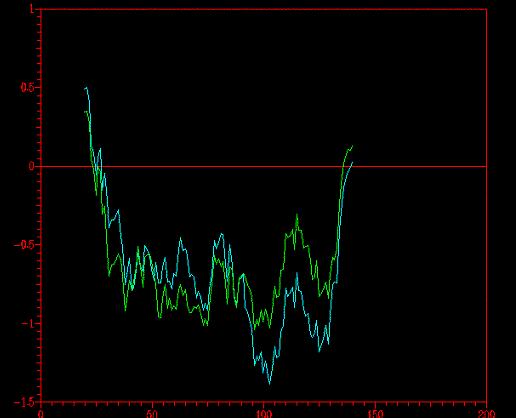
El programa PROSAII nos ofrece el perfil energético de la proteína. Considerando que la molécula debe tener una energia libre negativa para considerarse estable, todo aquello que esté por encima del umbral de 0 será considerado "sospechoso". De esta manera, podemos decidir cuál de los tres modelos es energéticamente más probable (aquel que tenga menos picos positivos en el gráfico).
Primeramente, comparamos los tres modelos obtenidos (floriA-1A-cutA)
y nos llevamos esta sorpresa.
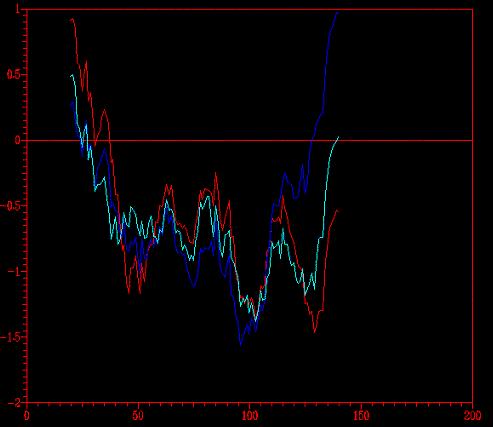
(*) En rojo tenemos
el primer modelo (con todos los templates), en azul oscuro el mod-1 ( sin
el 1emsA), y en azul claro el modcut (con el template 1emsA recortado).
En este gráfico vemos que la idea de obviar el 1emsA no fue brillante
(observense los picos positivos en el margen izquierdo y derecho del gráfico).
Sin embargo, modcut (en azul claro) tiene mejor perfil que mod1 (en
rojo).
* SEGUNDA PRUEBA (haciendo contactos...)
El paso siguiente será decidir cuál de las dos estructuras
que nos dio modcut (floricutA.pdby
floricutB.pdb)
es mejor. Para ello podríamos comparar sus perfiles energéticos.
Resultado: no eran significativamente diferentes, por tanto, debemos recurrir
al siempre dispuesto Proceck para escoger el mejor modelo.
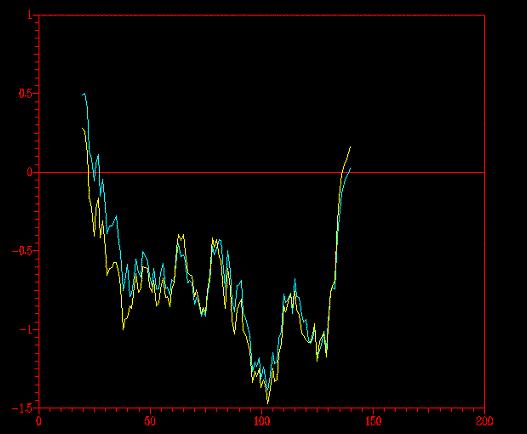
(*) En amarillo, floricutB.pdb y en azul claro floricutA.pdb
Utilizamos PROCHECK , un programa que comprueba si la disposición de los residuos es posible. Cuando encuentra algo que falla lo marca como "bad-contact" (especificados en los ficheros floricutA.sum y floricutB.sum). Así pues, el número de "bad-contacts" nos servirá para discriminar el modelo menos probable. El programa necesita saber con que resolución debe trabajar. Para ello, utilizará la mayor de entre totos los templates. Estas resoluciones eran:
* 1emsA:2,80
* 1kpaA: 2,00
* 1av5A: 2,00
* 2fit: 1,90
* 4fit: 2,50
* 4rhn: 1,90
* 6rhn: 2,15
Como en este caso la resolución mayor es 2,80 correspondiendo con
1emsA.
<<< P R O C H E C K S U M M A R Y >>>
floricutA.pdb 2.8 160 residues
* Ramachandran plot: 86.1% core 11.8% allow 1.4% gener 0.7% disall
* All Ramachandrans: 7 labelled residues
(out of 158)
Chi1-chi2 plots:
0 labelled residues (out of 102)
Main-chain params: 6 better
0 inside 0 worse
Side-chain params: 5 better
0 inside 0 worse
* Residue properties: Max.deviation:
4.0 Bad
contacts: 3
*
Bond len/angle: 5.6 Morris et al class:
1 1 2
G-factors Dihedrals: 0.00 Covalent: -0.32 Overall: -0.11
M/c bond lengths: 98.4% within limits
1.6% highlighted
M/c bond angles: 91.7% within limits
8.3% highlighted
Planar groups: 100.0% within limits
0.0% highlighted
<<< P R O C H E C K S U M M A R Y >>>
floricutB.pdb 2.8 160 residues
* Ramachandran plot: 88.9% core 6.9% allow 1.4% gener 2.8% disall
* All Ramachandrans: 7 labelled residues
(out of 158)
Chi1-chi2 plots:
1 labelled residues (out of 102)
Main-chain params: 6 better
0 inside 0 worse
Side-chain params: 5 better
0 inside 0 worse
* Residue properties: Max.deviation:
4.0 Bad
contacts: 5
*
Bond len/angle: 9.1 Morris et al class:
1 1 2
G-factors Dihedrals: 0.01 Covalent: -0.29 Overall: -0.10
M/c bond lengths: 98.9% within limits
1.1% highlighted
M/c bond angles: 91.5% within limits
8.5% highlighted 1 off graph
Planar groups: 100.0% within limits
0.0% highlighted
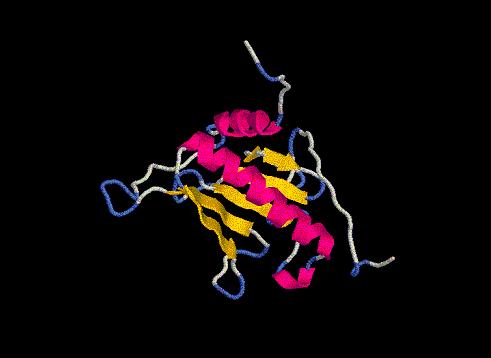
*AND THE WINNER IS.... el modelo floricutA.pdb por tener un menor número de bad contacts.
Una vez decidido cuál es el modelo, pasaremos a optimizarlo para intentar mejorar su perfil energético.
Para ello utilizaremos el programa GRUMOS y en concreto su opción "optimization", que encuentra aquellas conformaciones en que la energía de la molécula se sitúa en un mínimo. Después de muchas horas juntos, GRUMOS se rindió a nuestros encantos y nos dió una serie de ficheros por orden cronológico numerados del 1 al 15 en formato gsf. Este formato es parecido a pdb, pero no es visualizable por Rasmol.
No nos pongamos nerviosos, que para eso se inventó el programa XAM, que superpone estructuras: le intoducimos unas coordenadas y nos devuelve un archivo en formato pdb, en que a una de las estructuras le ha aplicado una matriz de rotación y de traslación que él solito ha creado. Superponemos el gsf-1 y el gsf-15 y obtenemos el fichero floricutAxam.pdb, al que rebautizaremos con el nombre de floricutAoptim.pdb, tras haberle eliminado las coordenadas del primer modelo optimizado, el gsf-1. Nos quedamos con las coordenadas que han resultado de la última optimización.
Este nuevo modelo optimizado será sometido de nuevo a la prueba
del PROSAII y comparado con el modelo floricutA.pdb.
(*) En el gráfico, en verde el modelo optimizado (floricutAoptim.pdb)
frente al no-optimizado en azul claro (floricutA.pdb).
Además,
lo pasamos por PROCHECK obteniendo:
<<< P R O C H E C K S U M M A R Y >>>
floriAoptim.pdb 2.8 160 residues
* Ramachandran plot: 75.7% core 22.2% allow 0.7% gener 1.4% disall
* All Ramachandrans:
13 labelled residues (out of 158)
* Chi1-chi2 plots:
4 labelled residues (out of 102)
Main-chain params:
6 better 0 inside
0 worse
Side-chain params:
5 better 0 inside
0 worse
Residue properties: Max.deviation:
4.5
Bad contacts: 0
Bond len/angle: 3.5 Morris et al class:
1 1 3
G-factors Dihedrals: -0.48 Covalent: 0.15 Overall: -0.23
M/c bond lengths: 99.9% within
limits 0.1% highlighted
M/c bond angles: 95.2%
within limits 4.8% highlighted
* Planar groups:
66.7% within limits 33.3% highlighted
9 off graph
¡ Han desaparecido los bad-contacts !!!!!!!!!!!!!!!!!!
Definitivamente
es un buen modelo, pero nosotras nunca tenemos bastante...
 ¡A
MOVER LOS LOOPS!
¡A
MOVER LOS LOOPS!
No os pongais nerviosos, que ya llegamos al final:
Ahora sólo nos queda someter a nuestro modelo al programa GRUMOS para hacer la dinámica molecular del modelo. Se trata de hacer una simulación en la que se analiza las distintas variaciones en el espacio de cada uno de los átomos (debidas a la flexibilidad de los enlaces, dihedros, ... etc.) de una proteína para ver como se mueve, es decir, su dinámica.
La opción de dinámica analizará las energías
de las conformaciones de los diferentes microestados de floricutA.pdb
y nos dará las coordenadas de sus átomos en formato gsf.
Le hemos pedido a Mr. Grumos que nos dé las coordenadas de 10 conformaciones,
para luego superponerlas mediante el programa XAM y ver qué partes
de nuestra estructura son las que más se modifican, y por tanto,
las menos estables. Esperamos, pues que el centro activo de la proteína
(una tríada de histidinas) se mantenga quietecito y que sean los
loops que no hemos podido modelar los que se muestren más bailongos:
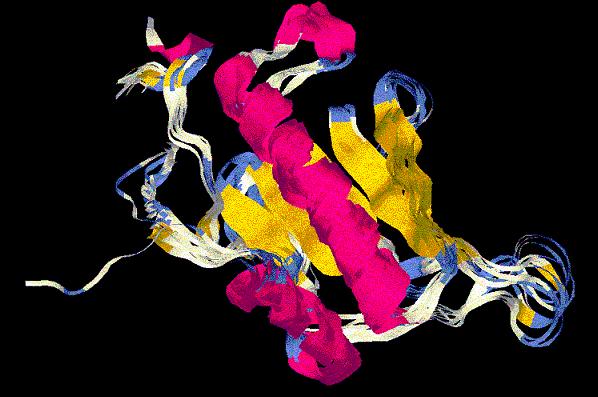
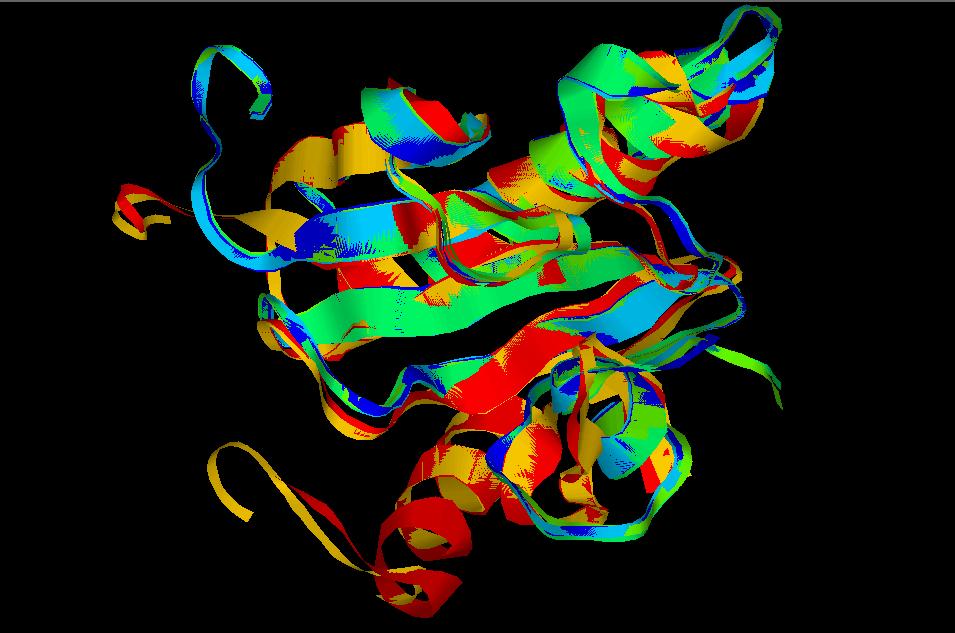
(*) En la imagen, la dinámica del modelo floricutA.pdb.

(*) Detalle de la imagen anterior, con las histidinas que conforman
el centro activo (residuos 111, 113 y 115) marcados en rosa. En esta
hendidura será donde se acoplará el sustrato.



 RIZANDO EL RIZO.
RIZANDO EL RIZO.