 LA
PRIMERA FLOR DE LA PRIMAVERA .
LA
PRIMERA FLOR DE LA PRIMAVERA .
Ya
tenemos los amiguetes estruturales de Floripondio. ¿ Qué
hacemos ahora con ellos?
Una vez descomprimidos los ficheros, el programa CLUSTALW utilizará
los fastas de los templates para darnos un alineamiento secuencial
de éstos (tempmod1.aln)
. Este formato clustalw es necesario para que el programa STAMP pueda crear
el alineamiento estructural (HIT.pdb).
Hemos de tener en cuenta que no queremos alineamiento por secuencia, por
lo que utilizaremos una opción de STAMP llamada alignfit. El fichero
resultante, al estar en formato pdb, es visualizable a través del
programa Rasmol, dándonos esta bonita imagen.
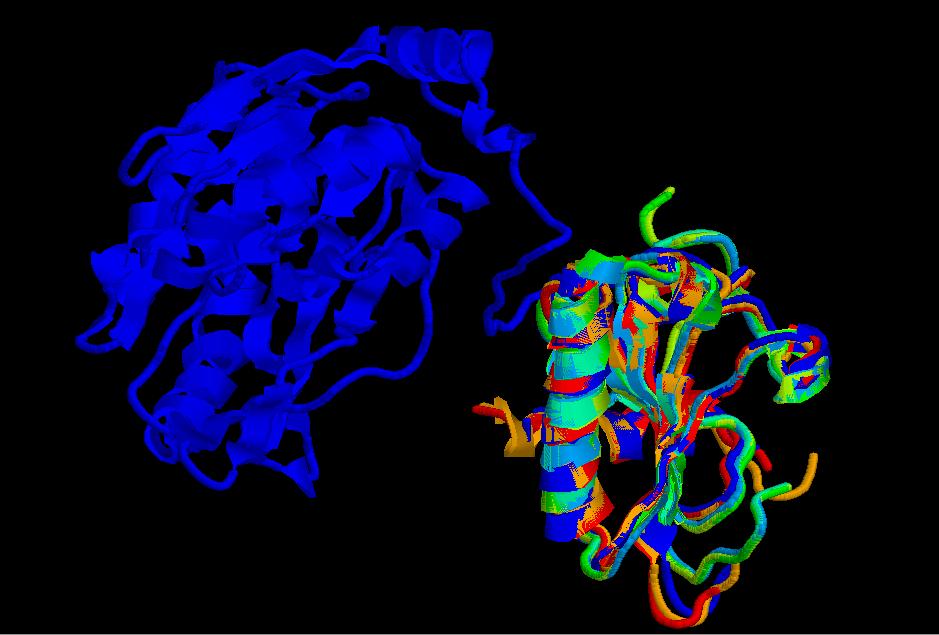
Como vemos en esta imagen, uno de los templates ( 1emsA, cadena azul) es más largo que los demás. Otra cosa que podemos observar es que la estructura consta de 2 hélices y 5 láminas, dominio típico de la super-família de las HIT proteins.
El siguiente paso será crear una nuevo matriz oculta de Markov, esta vez según la estructura (mod1.hmm) , mediante HMMER. Con esta matriz, y los fastas de los templates más el de Floripondio ( totmod1.fa) obtendremos el fichero tempmod1.ali, que será sobre el que trabajará el Modeller.
¡Estamos llegando al final! Ahora modificaremos el formato de totmod1.fa
hasta obtener un fichero de tipo pir (tempmod1.pir)
y crearemos las instrucciones del Modeller en el fichero mod.top.
En este fichero se indica qué matriz de Markov en formato pir se
utilizará y sobre qué templates. Ahora es cuando Modeller
entra en acción y nos da dos ficheros de tipo pdb (floriA.pdb y
floriB.pdb) visualizables por Rasmol.
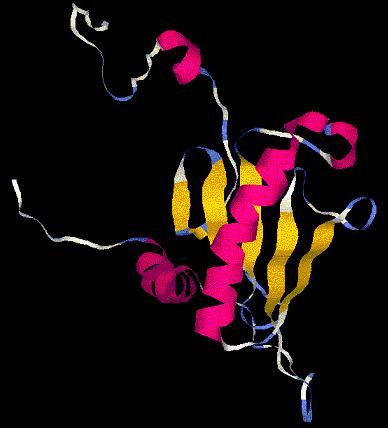
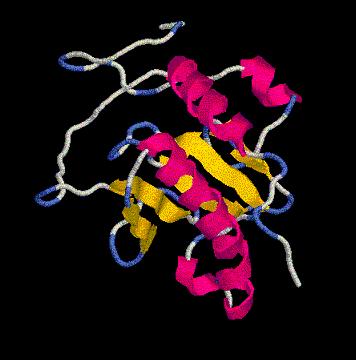
floriA.pdb
floriB.pdb
Las tres histidinas implicadas en el centro activo se localizan en los
residuos 111, 113 y 115.

(*) En la imagen, los tres residuos de histidina del centro activo marcados en verde.
Seguidamente, comparamos los perfiles energéticos de los dos
modelos obtenidos ( A y B) con el programa ProsaII, en el que no se aprecian
diferencias significativas.
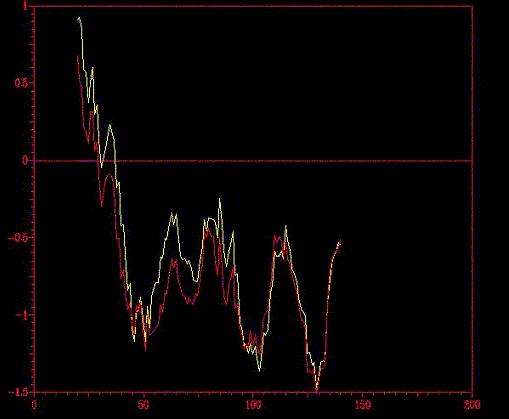
(*) En el gráfico,
floriA.pdb en amarillo y floriB.pdb en rojo
¡HEMOS
OBTENIDO EL PRIMER MODELO!


