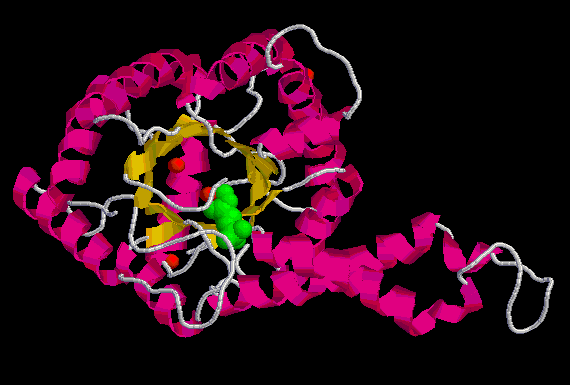
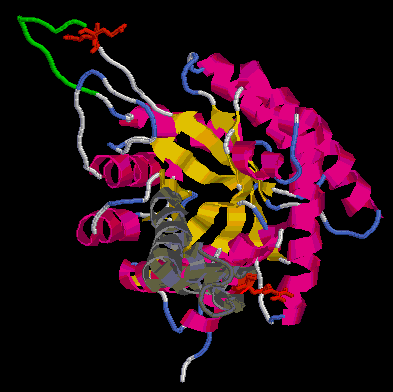
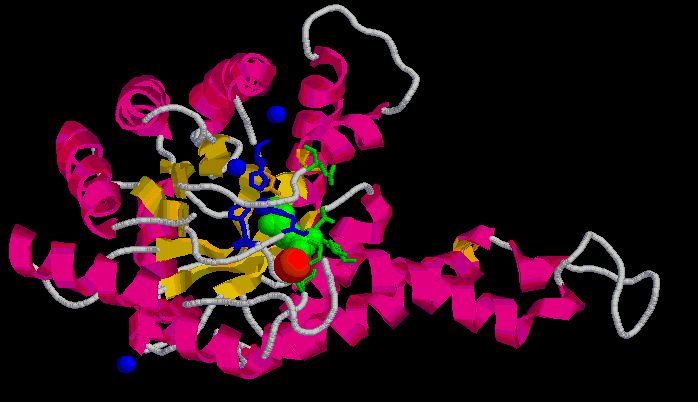
Figura 1. Estructura d'1b57 unida a PGH (verd) i Zn (vermell).
Ariadna Escalona & Carme Gubern
La proteïna a modelar es una fructosa bifosfat aldolasa (FBP-aldolasa) de classe II,de 343 aminoàcids, identificada al microorganisme Corynebacterium glutamicum.
Les fructoses bifosfat aldolases participen en dues grans rutes metabòliques: la gluconeogènesi (síntesi de fructosa bifosfat a partir d'hidroxiacetil fosfat, DHAP, i gliceraldehid 3-fosfat, G3P) i glicòlisi (catalitza la reacció inversa).
Les aldolases són dividides en dos grups, les de Classe I i les de Classe II, depenent majoritàriament del mecanisme de reacció. Tot i que es coneixen més les de Classe I, se sap que les de Classe II són més estables, i es creu que poden arribar a ser molt importants en la química de la biotransformació un cop el seu mecanisme de catàlisi i selecció siguin ben entesos. D'altra banda, en no trobar-se en mamífers, s'ha proposat que representin una diana per a nous fàrmacs antibacterians.
Les FBP-aldolases de classe II presenten una dependència absoluta
pels cations divalents, generalment Zn,
i són activades per cations monovalents. S'ha vist que comparteixen
un 15% d'homologia amb les de classe I.
ESTRUCTURA DE LES FBP-ALDOLASES DE CLASSE II
La primera estructura d'una aldolasa de classe II que va ser determinada va ser la de la proteïna fuculosa-1-fosfat d' E. coli. Posteriorment, es van modelar dues FBP-aldolases també d' E. coli que presentaven un plegament diferent al de la FucA: una estructura de barril (alfa/beta)8 homodimèrica. La nostra proteïna presenta aquesta mateixa estructura.
La subunitat és la clàssica (alfa/beta)8 observada en una gran quantitat de proteïnes. El dímer es manté estable mitjançant forces de Van der Waals i ponts d' Hidrogen que s'estableixen entre els residus que es troben al final de les hèlix alfa-10 i alfa-11 d'ambdues subunitats.
Tot i la gran variabilitat en la funció catalítica dels barrils (alfa/beta)8, la família de les FBP-aldolases comparteixen una localització i geometria comunes del centre actiu en una butxaca polar formada pels finals dels C-terminals de les làmines beta i els loops que les connecten amb l'extrem N-terminal de les hèlix alfa següents. Cada monòmer requereix la presència de, com a mínim, 1 àtom de Zn per la catàlisi. Aquest catió divalent juga un paper crític en l' estabilització de l'estat de transició del substrat durant la reacció.
Un estudi d'unió d'un anàleg de l'estat de transició del substrat fisiològic, el PGH (fosfoglicolohidroxamat), a la proteïna 1b57 d' E. coli (una FBP-aldolasa de classe II) ha permès determinar els residus importants tant a nivell estructural com funcional que es descriuen a continuació.
Descripció estructural d'1b57:
Figura 1. Estructura d'1b57
unida a PGH (verd) i Zn (vermell).
Presenta un total de 8 làmines beta i 11 hèlix alfa. El seu barril (alfa/beta)8 està format per les làmines beta 1-8 més les hèlix alfa 2, alfa 4-9 i alfa 11. L'hèlix alfa 1 cobreix l'extrem N-terminal del barril, l'hèlix alfa 3, molt curta, uneix la làmina beta 2 amb l'hèlix alfa 4 i per últim, l'hèlix alfa 10 va des de la beta 8 a una regió loop.
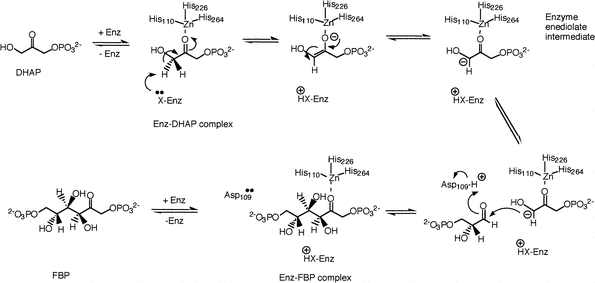
Els residus clau pel cicle catalític de les FBP-aldolases de classe II d' E. coli són els següents. La Thr289, la Ser267, la Gly227 i l' Asp288 s'ha vist que formen ponts d'hidrogen amb el grup fosfat del DHAP a través dels seus grups amino, i l' Asp109, la Ser61 i l'Arg331 són importants per la unió al G3P. Es creu que la Ser61 forma ponts d'hidrogen amb el grup fosfat del G3P, que l'Asp109 polaritzaria el C1 del G3P i l'Arg331 estaria implicada en la unió al fosfat del C6 del G3P de l'altre monòmer. Aquesta arginina es troba situada en l'hèlix 11, una de les dues implicades en la dimerització. Quan el DHAP i el Na s'uneixen a l'enzim, es produeixen tota una sèrie de canvis conformacionals al centre actiu que fan que el ió catalític de Zn passi d'una posició enterrada a una posició més superficial i exposada, que li permet unir-se al DHAP. Aquests canvis conformacionals són conseqüència de la rotació de les His110, 226 i 264, que són els lligands del Zn. El mecanisme de la reacció es pot veure aquí.
S'han detectat canvis conformacionals en els loops beta5-loop-alfa7,
beta6-loop-alfa8
i beta7-loop-alfa9. El loop beta5-loop-alfa7 sembla ser el més
dinàmic dels tres i per tant, tindria importància a nivell
funcional. Sembla que aquest loop cobreix el centre actiu després
d'unir-s'hi els substrats. Al final del C-terminal de la beta5 hi ha un
Glu que està conservat en la família de de les FBP-aldolases
de classe II que, a la 1b57, es tracta del Glu174,
que és un dels lligands del Zn en la seva posició més
enterrada. Dos altres Glu que es troben al loop, el Glu181
i el Glu182
en 1b57, s'ha vist que són acostats al centre actiu gràcies
al moviment de tancament del loop i interaccionarien de la següent
forma: el Glu181 ho faria amb l'àtom de Zn i el 182 amb el
DHAP.
Per trobar les seqüències homòlogues a la de la P19537, es va fer un PSI-BLAST contra la base de dades SWISSPROT; hi va haver convergència al 3er round. Les seqüències que es van escollir perquè tenien un valor de E-value baix i un percentatge d'homologia elevat van ser les següents:
Q9X8R6
ALF-STRCO
069600
ALF-MYLLE
P11604
ALF-ECOLI
P53818
ALF-CAMJE
051401
ALF-BORBU
Es van aliniar amb CLUSTALW i aquest aliniament es va utilizar per elaborar un model de Hidden Markov que, un cop calibrat, llençaríem contra la base de dades PDB per trobar els models estructurals que s'ajustessin millor amb la nostra proteïna. D'aquest hmmsearch contra el pdb es van trobar tres templates: l'1dosA, l'1b57A i l'1zen. Com que n'havien sortit només aquests tres, es va intentar trobar-ne de nous a través de la base de dades PFAM. Es va agafar tota la família de proteïnes dels templates que hi havia a pfam (90 seqüències en total), es van aliniar amb clustalw i es va construir un model Hidden Markof a partir d'aquest aliniament. De nou, es va fer un hmmsearch contra el pdb. No se'n van trobar de nous.
A partir d'aquest punt es va decidir elaborar dos tipus de models, un basat
en un aliniament de seqüència i l'altre basat en un
aliniament
d'estructura. Per fer el primer aliniament, es va fer un hmmalign
de les seqüències en format fasta dels templates i la de la
proteïna problema utilitzant el primer model
Hidden Markov
obtingut.
Per fer el segon tipus d'aliniament, es va fer un STAMP dels tres templates
i es va construir una matriu a partir d'aquest mitjançant
hmmbuild.
Aquest model de HiddenMarkov es va utilizar per fer l'hmmalign
de les seqüències en format fasta dels templates i la de la
seqüència problema.
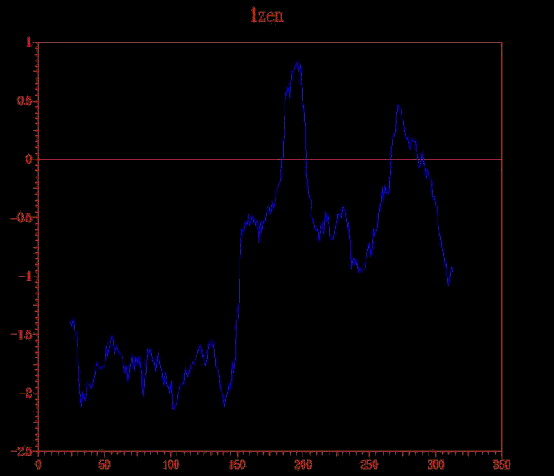
En analitzar els dos aliniaments obtinguts, es va observar que el template 1zen no aportava informació útil per modelar la proteïna problema ja que obria molts gaps a la seqüència problema. Per acabar d'estar-ne segurs, es va fer un PROSAII d'aquest template que confirmava que no era útil per construir un bon model, ja que presentava zones amb energies positives. Això no passava ni amb 1dosA ni amb 1b57A com es pot veure aquí. Es va decidir, doncs, eliminar-lo dels aliniaments i es va repetir tot el procés per obtenir els aliniaments estructural i de seqüència definitius.
Per tal de determinar si calia fer alguna modificació en els aliniaments, es va fer una predicció d'estructura secundària a partir de la seqüència en format fasta de la proteïna problema. Afortunadament cap dels gaps presents als aliniaments es trobaven enmig d'una hèlix alfa o làmina beta i els residus importants per la reacció catalítica tampoc estaven afectats; així que no es va fer cap modificació.
Arribat a aquest punt, es va procedir a executar el MODELLER obtenint 2 models a partir de cada aliniament. S'escollí el segon model basat en l'aliniament de seqüència perquè era el que presentava el millor patró energètic en l'anàlisi de prosall (totes les energies eren negatives). Afegim que en la superposició de tots quatre models mitjançant stamp no es van observar diferències significatives. Es va procedir a fer el PROCHECK, amb una resolució de 2 A, d'aquest model basat en seqüència i dels templates. Seguidament es va córrer la topologia i l'optimització amb GROMOS per millorar els resultats del procheck, de manera que el model final va ser l'optimitzat. Al model optimitzat se li va fer un anàlisi utilitzant prosaII i procheck; a més, es determinaren els canvis conformacionals processant la dinàmica i el patró de B-factors processant l'anàlisi, també amb gromos. (Per problemes tècnics en córrer la dinàmica, es va haver d'eliminar la prolina que té la P19537 al principi de la seqüència).
Paràmetres utilitzats a gromos:
Finalment es va superposar el lligand PGH de 1b57A al model optimitzat
mitjançant stamp amb l'objectiu d'analitzar si en el model es mantenia
la geometria del centre actiu.
Model no optimitzat Model optimitzat
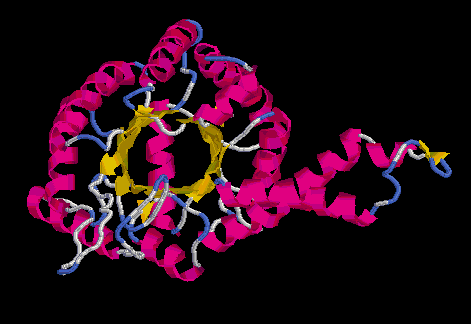
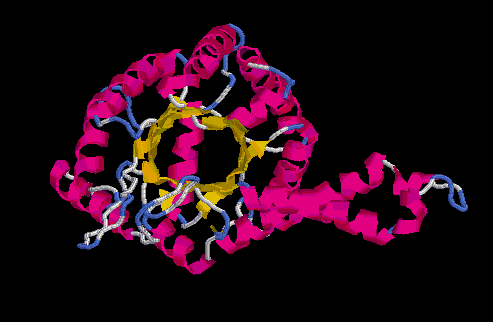
Superposició dels prosaII dels dos models:
Resultats del procheck: model
no optimitzat/optimitzat
templates
Resultats
Dinàmica B-factor
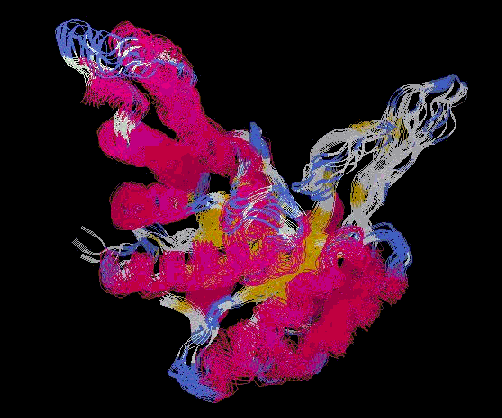
Figura5. Superposició
amb xam de la dinàmica feta amb gromos.
NOTA: La discussió s'ha fet prenent com a model el template 1b57, ja que és l'únic que té unit el lligand.
PROSAII
Tal i com s'observa a la figura 4 , s'ha produït una disminució d'energia en diferents zones del model amb l'optimització, per tant, una millora energètica. Aquesta millora s'ha donat especialment a l'extrem C-terminal del model. En aquesta zona, abans de l'optimització, l'energia tenia valor positiu. Aquesta regió correspon aproximadament amb les hèlix alfa 10 - 11, segons la numeració referent a 1b57, o hèlix alfa 13 -14, per la numeració del model.
L'important es veure quin patró energétic segueix el nostre model, respecte al dels templates, una vegada optimitzat. Comparant amb els templates considerarem les tres zones que es mostren a la següent figura encerclades.
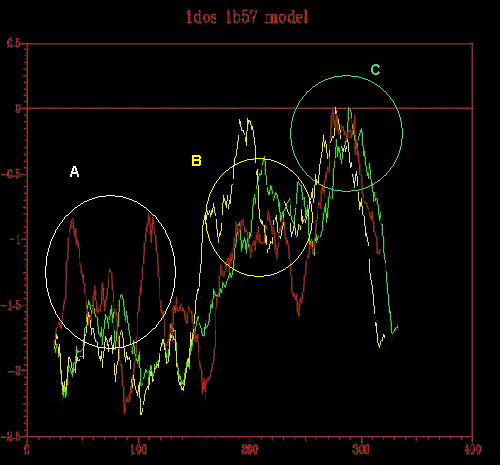
Figura 6. ProsaII
dels templates (1b57 i
1dos)
i el
model.
A) En aquesta zona s'han analitzat els dos pics situats entre 1-60 residus i 90-140 residus.
B) Regió de180-250 aminoàcids, on el model optimitzat té un nivell energètic inferior al dels templates.
C) No
hi ha diferència entre templates i models en la zona compresa entre
els
aminoàcids 260-340.
PROCHECK
Amb
l'optimització del model s'ha aconseguit reduir a 0 els bad contacts
que presentava el model no optimitzat (3). Tot i això ha augmentat
a un 1.4% la proporció de residus situats en regions no permeses.
Pel que fa al percentatge de residus situats al core i a les zones permeses,
no s'observen diferències significatives entre el model optimitzat
i els templates. On sí que hi ha diferències entre templates
i model és a les zones no permeses: als templates 0% i al model
optimitzat 1.4%. Mapes de Ramachandran
.
DINÀMICA
Amb la dinàmica explorem l'espai
conformacional del nostre model optimitzat. El que ens mostra són
les diferents conformacions que poden adoptar les estructures (hèlix
alfa, làmines beta o loops) en l'estat final d'energia després
de l'optimització. Els motius més ben definits, com pot ser
el barril (alfa/beta)8 de la nostra proteïna, seran els més
estables i els que estaran millor modelats; per tant, a la dinàmica
observarem una mínima mobilitat. En canvi, hèlix interrompudes,
loops llargs, etc. presentaran més mobilitat en no ser tan estables.
El modeller només proposa un únic tipus de conformació,
cosa que no vol dir que no n'existeixin d'altres que s'adiguin també
amb l'estat energètic proposat a l'optimització per cada
zona. La dinàmica, doncs, el que fa és mostrar les altres
possibilitats conformacionals viables en cadascuna d'elles.
Amb B-factor es defineix el patró de temperatura dels residus
que formen la proteïna. A major temperatura, major mobilitat.
A la figura 5 veiem el resultat de la dinàmica
aplicada al nostre model. S'observen dues regions amb el major grau de
mobilitat: una correspon al loop beta5-loop-alfa7, el més
llarg de la proteïna, i que participa en la catàlisi enzimàtica,
a través dels glutàmics 166 i 167, i l'altra correspon a
un loop més petit, el hèlix13-loop-hèlix14;
totes dues hèlix estan interrompudes. D'altra banda, en analitzar
el gràfic dels B-factors es poden apreciar
dos pics importants: un situat entre els residus
167 i 177 i l'altre entre els residus
273 i 321, que es corresponen aproximadament
amb els loops beta5-loop-alfa7 i hèlix13-loop-hèlix14,
respectivament.
Puntualitzar també
que a la figura 5 s'observa mobilitat per al loop beta6-loop-alfa8, així
com un pic, menor, situat entre els residus 209 i 250, on es situa aquest
loop que conté la histidina 210.
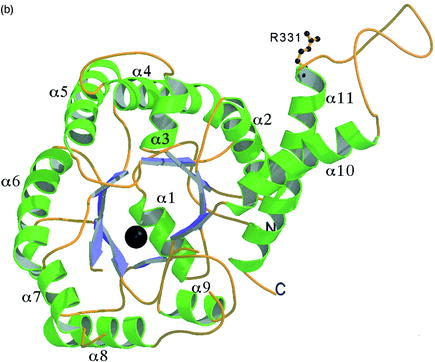
A la imatge veiem la situació dels Glu
166 i Glu 167 al loop beta5-loop-alfa7,
que corresponen al template 1b57 als Glu181 i 182. A 1b57 s'ha vist
que ambdos glutàmics són acostats al
centre actiu gràcies al moviment de tancament del loop i interaccionarien
de la següent forma: el Glu181 ho faria amb l'àtom de Zn i
el 182 amb el DHAP. Aquest fet concorda tant amb el pic de fluctuació
observat al gràfic com amb el moviment en la superposició
de la dinàmica.
També veiem la situació
de l'Arg 312
a l'hèlix 14 del loop alfa13-loop-alfa14
(Arg331
al template) que s'uneix
al fosfat del C6 del G3P
de l'altre monòmer. Les hèlix 13 i 14 són les
que estableixen forces de Van-der-Waals i ponts d'Hidrògen amb les
hèlix alfa 13-14 del monòmer veí en la dimerització.
Seria d'esperar una certa flexibilitat en aquesta zona per què les
4 hèlix dels monòmers estableixin les interaccions adequades
per a la dimerització. Aquesta flexibilitat es veu reflexada en
la superposició de la dinàmica i al pic del gràfic
de B-Factor, tot i que considerem que és massa elevat.
Finalment,
afegim que la diferència en mobilitat dels loops beta5-loop-alfa7
i beta6-loop-alfa8 concorda amb el que es va veure en l'estudi d'unió
1b57-PGH: el beta5-loop-alfa7 és el més dinàmic.
EL MODEL MANTÉ LA MATEIXA FUNCIONALITAT QUE EL TEMPLATE 1B57?
Aquesta pregunta la podem respondre comprovant que les distàncies entre els residus que formen el centre actiu del template i el substrat es mantenen també al nostre model.
Per observar la conformació que adopten els residus del centre actiu
de la proteïna 1b57 unida al seu substrat, el PGH, i al Zn, cliqueu
aquí.
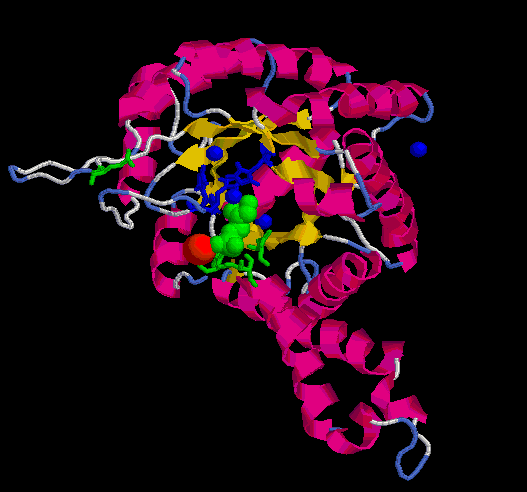
En verd s'hi ha representat la molècula de PGH i els aminoàcids
que interaccionen amb el seu grup fosfat; en blau, els àtoms de
Zn i les histidines que hi interaccionen; en vermell, l'àtom de
Na i, finalment, en taronja els residus que interaccionarien amb l'altre
substrat, el G3P (no s'ha trobat cap template que estigués unit
als dos substrats). Si s'observa detingudament la imatge, es veu com les
tres histidines estableixen interaccions de "stacking" a través
dels seus anells aromàtics i l'àtom de Zn més proper
al substrat. També es pot apreciar com els residus en verd (thr289,
ser267, gly227i asp288) podrien formar ponts d'hidrògen entre els
seus grups amino i el grup fosfat del PGH. El glu182, tot i estar massa
separat en la imatge (com ja s'ha dit, es troba al loop entre la beta 5
i l'alfa 7), també hi interaccionaria un cop el moviment de tancament
del loop hagués finalitzat.
Per simplificar l'anàlisi de conservació de la funcionalitat, només s'han mesurat les distàncies entre les histidines i l'àtom de Zn més proper al substrat i les distàncies entre el grup fosfat del substrat i els grups amino de cadascun dels residus que hi interaccionen.
Al template, aquestes distàncies són les següents:
| HISTIDINA ( grup ND1) | 110 | 226 | 264 |
| ZN | 4.205 A | 1.924 A | 3.866 A |
| grup N | THR 289 | SER 267 | GLY 227 | ASP 288 | GLU 182 |
| P | 3.779 A | 3.844 A | 3.857 A | 3.750 A | 9.439 A |
Al model, tots els residus que formarien part del centre actiu d'una FBP-aldolasa es troben conservats. Si s'observa la imatge de la P19537 unida al PGH (que hauria de simular, com en el cas del template, l'estat de transició del DHAP, el seu suposat substrat fisiològic), es pot apreciar la presència dels mateixos residus. Si es compara la conformació que adopten amb la del template, s'observa com els tres residus conservats d'histidina -essencials per a al manteniment de la geometria trigonal de l'àtom de Zn- no es troben tan ben col·locats entorn a l'àtom de Zn més pròxim al PGH, cosa que indicaria que les interaccions de "stacking" entre els seus respectius anells i el catió divalent no són tan fortes com al template. Per contra, si s'observa la conformació dels residus thr275, ser253, gly252 i asp 274 (corresponents als residus thr289, ser267, gly 227 i asp288 del template), s'aprecia que sí que adopten una conformació que podria ser correcta per què els seus respectius grups amino interaccionessin amb el fosfat del PGH.
Les distàncies trobades al model són les següents:
| HISTIDINA (grup ND1) | 94 | 210 | 250 |
| ZN | 4.182 A | 4.786 A | 2.673 A |
| grup N | THR 275 | SER 253 | GLY 252 | ASP 274 | GLU 167 |
| P | 3.256 A | 2.905 A | 4.060 A | 3.929 A | 20 A |
Una puntualització que s'ha de fer respecte a aquestes distàncies és que són massa grans per què s'estableixin interaccions tant de "stacking" com per què es formin ponts d'hidrogen. Aquest fet segurament és degut a la forma que té la versió de MODELLER utilitzat de representar les cadenes laterals dels residus: només escull una única posició per la cadena lateral de cada aminoàcid, que pot ser que no sigui l'adequada perquè es mantinguin les distàncies corresponents i es puguin establir les interaccions corresponents. Això no significa, doncs, que no s'hagin identificat bé els residus del centre actiu tant del template com del model.
Respecte a les distàncies entre les tres histidines i l'àtom de Zn, s'observa que no es mantenen excepte en el cas de les histidines 110 i 94 respectivament. Per contra, respecte a les interaccions amb el grup fosfat del substrat, sembla que s'hagin mantingut les distàncies en els casos de la glicina i aspàrtic però no en els casos de la treonina, la serina ni el glutàmic.
Amb aquesta informació podem afirmar que la nostra proteïna
manté, aproximadament, la conformació del centre actiu que
s'ha trobat a 1b57. Pensem, però, que s'hauria de disposar d'altres
templates de la família units als dos substrats (DHAP i G3P) per
disposar de més dades per acabar de definir la disposició
espacial del seu centre actiu.
Després d'analitzar tots els resultats obtinguts podem concloure que el model de la proteïna P19537 manté la topologia de les FBP-aldolases de classe II Zn depenents: barril (alfa/beta)8. Considerem que el model final podria ser funcional per les següents raons:
- Conservació de residus implicats en unió a Zn (His94,210,250 i Glu166), DHAP(Thr275, Ser253, Gly252, Asp274 i Glu167) i G3P (Arg312) a les mateixes estructures que al template.
- S'han observat canvis conformacionals en els loops determinants
per la catàlisi enzimàtica:
beta5-loop-alfa7: Conté els Glu166
i 167 que interaccionarien amb el Zn i el DHAP, respectivament, en ser
acostats al centre actiu gràcies al moviment de tancament del loop.
Així com al template, és el loop més
dinàmic.
beta6-loop-alfa8: Conté la His210. La conformació d'aquest loop en el model fa que no es mantinguin les distàncies entre aquest residu i el Zn. Però els resultats de la dinàmica indiquen que presenta certa mobilitat i, per tant, podria interaccionar correctament amb el catió.
alfa13-loop-alfa14: Encarregades de la
dimerització. A l'hèlix 14 es conserva Arg312 que interacciona
amb el G3P del monòmer veí. El model respecta la disposició
espacial de les hèlix necessària per la dimerització.
Simultàniament, la informació de la dinàmica mostra
flexibilitat de les hèlix per afavorir les interaccions pertinents.
- Conservació
de les distàncies entre residus al centre actiu: hi ha distàncies
que estàn conservades; respecte a les no conservades, sabem que
alguns residus es troben en zones mòbils i que per tant podrien
acabar interaccionant.
COM PODRIEM MILLORAR EL MODEL?
Sobre el model presentat faltaria fer, per exemple, un anàlisi de la dinàmica dels templates i comparar-lo amb l'obtingut a partir del model per determinar si cal remodelar algun dels loops del centre actiu.
Considerant que la proteïna 1b57 és la que ha donat els resultats
més bons al procheck i és sobre la que hem trobat més
informació respecte als seus lligands i els canvis conformacionals
que hi tenen lloc, una posibilitat seria eliminar l'altre template,
l'1dos, de l'aliniament clustalw tant d'estructura com de seqüència
i repetir el procés. Segurament haguéssim vist millores tant
al procheck com al prosaII, i després de superposar el model optimitzat
amb el lligand de l'1b57, les distàncies del centre actiu respecte
als lligands estarien més conservades.
AGRAÏMENTS
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}