

![]()
Abstract
Introducció
Objectius
Metodologia:
1.-
Obtenció de la seqüència problema
2.-
Obtenció dhomologies:
2.1.- Homologia a nivell de seqüència
2.2.- Homòlegs estructurals
2.2.1.- Anàlisi de les proteïnes
2.2.2.- Superposició destructures
3.-
Obtenció del perfil i aliniament
4.-
Modelat de la proteïna
4.1.- Prosa II
4.2.- Procheck
5.-
Optimització del model
5.1.- Modelatge dels loops
5.2.- Minimització denergies
6.-
Estudi de la Dinàmica Molecular
Resultats:
1.-
Obtenció de la seqüència problema
2.-
Obtenció dhomòlegs
2.1.- Homologia a nivell de seqüència
2.2.- Homòlegs estructurals
2.2.1.- Superposició destructures
3.-
Obtenció del perfil i aliniament amb el HMM
4.-
Modelat de la proteïna
5.-
Optimització del model
5.1.- Modelatge dels loops
5.2.- Minimització denergies
6.-
Estudi de la Dinàmica Molecular
Discussió
Bibliografia
PROGRAMA DE
RECONEIXEMENT DE MOTIUS D'UNIÓ A ATP
ABSTRACT
La relació entre els nucleòtids (codons) i els aminoàcids
sestableix mitjançant les reaccions que porten a terme les aminoacil-tRNA
sintetases. Aquests enzims uneixen els seus substrats ATP, aminoàcids
i tRNA- i estabilitzen els estats de transició en la reacció
daminoacilació.
En aquest treball sha modelat lestructura tridimensional de la tirosil-tRNA
sintetasa dAquifex aeolicus i shan analitzat les seves característiques
estructurals i funcionals.
El treball inclou el disseny dun programa que permet la identificació
de totes les molècules que continguin un motiu dunió a ATP,
entre les quals es troben les aminoacil-tRNA sintetases.
La traducció fidel del codi genètic depèn de la unió correcta dels aminoàcids als seus RNAs de transferència corresponents. Aquesta reacció està catalitzada per les aminoacil-tRNA sintetases, una família de 20 enzims. Hi ha un enzim diferent per cadascun dels 20 aminoàcids i com que el codi és degenerat (61 codons per 20 aminoàcids), cada sintetasa pot reconèixer i aminoacilar cadascun dels diferents tRNA isoacceptors que es corresponen amb laminoàcid determinat.INTRODUCCIÓ
La reacció, anomenada daminoacilació o càrrega
dels tRNAs, es porta a terme en dos passos. En primer lloc, laminoàcid
és activat quan ataca una molècula dATP, donant lloc a un
intermediari, laminoacil-adenilat, i a pirofosfat inorgànic. En
segon lloc, laminoàcid és transferit a la ribosa terminal
3del tRNA corresponent, donant lloc a laminoacil-tRNA i a AMP.
Classes i subgrups
La funció que realitzen les aminoacil-tRNA sintetases és comú per totes les proteïnes daquesta família, però són extremadament diverses pel que fa al seu tamany molecular, estructura quaternària i seqüència. La comparació de les seves seqüències conjuntament amb el seu anàlisi tridimensional, va permetre descobrir algunes característiques comunes que divideixen la família en dues classes (5). Laminoacil-tRNA sintetasa modelada en aquest treball és la tirosil-tRNA sintetasa dAquifex aeolicus i pertany a la classe I.
Les aminoacil-tRNA sintetases de classe I tenen els motius de seqüència
conservats HIGH i KMSKS. El nucli catalític té una estructura
similar al plegament de Rossmann dunió a
nucleòtids (1). Els dos motius conservats formen part del domini
dunió a ATP (fig.1).
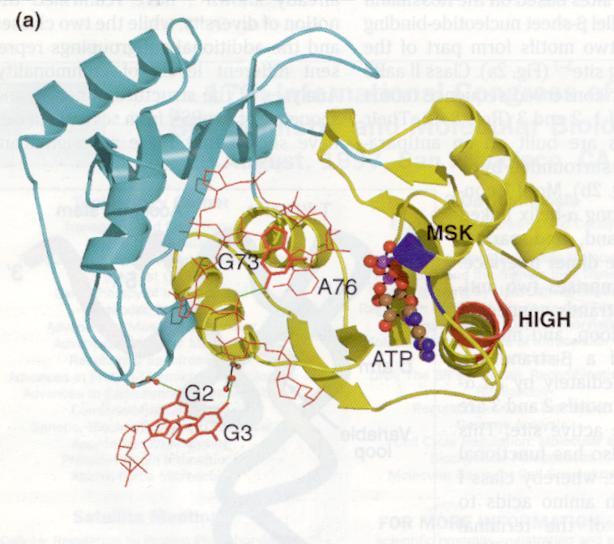
Fig 1. Domini catalític de les aminoacil-tRNA sintetases de classe I.
En aquest cas es tracta de la GlnRS. LATP i el lloc acceptor del tRNA corresponent estan en vermell. La localització dels motius característics
està indicada de la manera següent: MSK (blau fort), i HIGH (vermell).
Les aminoacil-tRNA sintetases malgrat ser molt diverses en estructura
quaternària, presenten alguns patrons. Les sintetases de classe
I són principalment monomèriques, excepte la TyrRS i la TrpRS
que són dímers obligats. Les sintetases de classe II tendeixen
a ser dímers.
|
|
Classe I |
Classe II |
| Motius conservats | HIGH, KMSKS | Motiu 1, 2 i 3 |
| Plegament del lloc actiu | Làmina beta paral·lela (plegament de Rossmann) | Làmina beta antiparal·lela |
| Lloc dunió al tRNA | Solc menor | Solc major |
Taula 1. Principals característiques diferencials de les dues classes daminoacil-tRNA sintetases.
També shan trobat característiques comunes addicionals
entre alguns membres de cada classe, i això ha permès la
divisió de cada classe en subgrups. Els subgrups no només
depenen de la presència de mòduls comuns, també estan
relacionats amb el tipus daminoàcid que les sintetases transfereixen
al tRNA. La tirosil-tRNA sintetasa conjuntament amb la triptofanil-tRNA
sintetasa pertanyen al grup dels enzims que carreguen aminoàcids
aromàtics als tRNAs.
Shan identificat per homologies de seqüència proteïnes
semblants a les sintetases. Aquestes proteïnes consisteixen principalment
en un sol domini de les aminoacils, la qual cosa reforça la idea
que les sintetases tenen un disseny modular. Per tant, les sintetases són
el resultat de lensemblatge de vàries peces al llarg de levolució.
És lògic que per aquest motiu les empremtes daquests
enzims apareguin en llocs insospitats. Un exemple daquestes homologies
de seqüència lil·lustra un tipus de citoquina que sassembla
molt al domini C-terminal de la tirosil-tRNA sintetasa de mamífers.
Sassembla fins a tal punt que tallant aquest extrem de la TyrRS sobserva
la mateixa activitat que la citoquina (3 i 4).
OBJECTIUS
Els objectius daquest treball es poden resumir en 2 punts molt
concrets:
- Modelatge de la tirosil-tRNA sintetasa a partir de proteïnes homologues. Daquesta manera es pretén determinar lestructura tridimensional de la tirosil-tRNA sintetasa dAquifex aeolicus, identificar-ne llurs dominis estructurals i funcionals, analitzar-ne la relació estructura-funció, i estudiar les semblances i diferències amb daltres tirosil-tRNA ja modelades.
- Disseny dun programa que permeti identificar les característiques
del domini dunió a ATP. Això ens serà útil,
ja que, com sha dit, la tirosil-tRNA sintetasa té un domini dunió
a ATP, el qual és imprescindible per dur a terme la seva funció.
METODOLOGIA
A continuació es detallen els passos i el procediment seguits
en lobtenció de lestructura tridimensional de la tirosil-tRNA
sintetasa dAquifex aeolicus, la qual sanirà modelant per tal dobtenir
una estructura el més optimitzada possible.
1.- Obtenció de Seqüència
Per a lobtenció de la seqüència en format FASTA de la tirosil-tRNA sintetasa dAquifex aeolicus (O67632) sha recorregut a la base de dades Swissprot.
2.- Obtenció dhomologies
2.1.- Homologia a nivell de seqüència
Blast al Swissprot per tal de trobar homologies de seqüència i alineament amb Clustalw.
2.2.- Homòlegs estructurals
Blast a la base de dades PDB, per tal dobtenir homòlegs estructurals de la tirosil-tRNA sintetasa dAquifex aeolicus. Sescullen els que presenten E-values més petits i, per tant, més homologia estructural, juntament amb alguns altres que presentin valors més elevats per tal que, un cop es faci la superposició destructures, sobtingui una superposició amb el mínim de regions que presentin buits o gaps estructurals.
2.2.1. - Anàlisi de les proteïnes
Es recorre a la base de dades SCOP per conèixer algunes característiques de les proteïnes homòlogues com la família a la qual pertanyen, etc. Això permetrà reduir laleatorietat a lhora de triar els pdbs.
2.2.2.- Superposició destructures
Es sobreposen els 7 pdbs homòlegs mitjançant el Stamp per tal de determinar el grau de semblança estructural a través de la visualització de les estructures amb el Rasmol.
3.- Obtenció perfil i alineament
Es crea un perfil de HMM (Hidden Markov), per cadascun dels grups estructurals diferenciats que ha mostrat la superposició amb lStamp. Es determina lScore de semblança entre el perfil i la seqüència problema, la qual cosa permet saber quin dels dos grups estructurals és més semblant a la proteïna problema. I salinea la proteïna problema amb el perfil més semblant a la tirosil-tRNA sintetasa dAquifex aeolicus, és a dir, el perfil més apte per modelar la proteïna problema. De lalineament resultant es talla lextrem C-terminal, ja que no és informatiu.
4.- Modelat de la proteïna
Sobté una primera aproximació del model de la tirosil-tRNA
sintetasa dAquifex aeolicus mitjançant el programa Modeller.
Sanalitzen lestabilitat de lestructura en funció de:
4.1.- Lespectre denergies mitjançant Prosa II. Aquest programa, basat en lanàlisi de potencials estadístics, permet saber quin dels models obtinguts és millor quant a lestabilitat energètica.
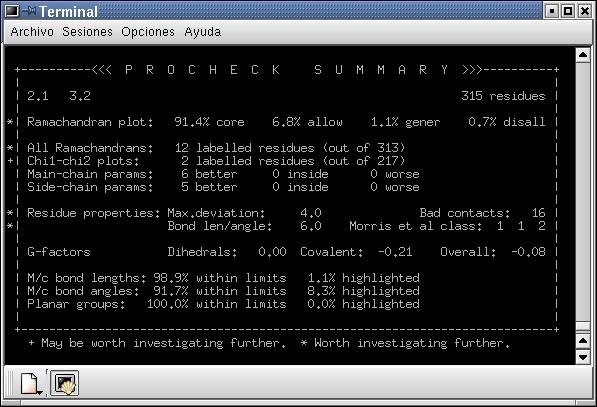
4.2.- Els angles, mitjançant Procheck. Sobté un mapa de Ramachandran (i una taula) on shi poden observar el % daminoàcids que, segons els angles phi i sigma, són correctes (core), permesos (allow), generosament permesos (gener) o dissolts (disall o no permesos). També informa sobre el nombre de bad contacts (o interaccions no permeses), entre daltres.
5.- Optimització del model
5.1.- Modelatge dels loops
Un cop obtingut un primer model es determina lexistència de loops i es modelen utilitzant la base de dades http://ibb.uab.es/loops
5.2.- Minimització d'energies
La minimització denergies es fa mitjançant Grumos. Se superposa mitjançant el Xam el primer i lúltim model per tal dobtenir un format que el Prosa II i el Procheck puguin reconèixer posteriorment. Sanalitza lestabilitat estructural, a partir de lespectre energètic i els angles, dels models optimitzats amb el Grumos.
6.- Estudi de la Dinàmica Molecular
A partir del model optimitzat amb el Grumos es fa un estudi de
la Dinàmica Molecular amb lopció Dynamic del Grumos.
Aquest estudi permet analitzar la flexibilitat de la proteïna, a base
dexplorar tot lespai conformacional possible.
RESULTATS
1.- Obtenció de la seqüència
problema
La seqüència de la tirosil-tRNA sintetasa dAquifex aeolicus, en format FASTA és la següent:
>sp|O67632|SYY_AQUAE Tyrosyl-tRNA synthetase (EC 6.1.1.1) (Tyrosine--tRNA
ligase) (TyrRS) - Aquifex aeolicus.
MTPEEQLRIIKEGTVEIIEEEELLKKLKEGRPLRVKAGFDPTAPDLHLGHVVLLQKLRQFQQLGHEVFFIIGDFTAMIGDPTGRSQTRPPLSREQVLENAKTYEHQVFKVLIPEKTTVVFNSTWLEELGTKGLIELCAKYTVARMLEREDFSKRFKEGIPIYIHEFIYPLLQAYDSVAIKADVEIGGTDQKFNLLIGRDIQREYGQEPQVCITLPLLVGTDGVRKMSKSYGNYVGITEDPKTMFAKIMSIPDEIMWDWFLLLTDYNKEEIEKMRREMHPMEAKKLLAFTIVKRFHSEEEARKAKEWWEKTFSQREFPEDAPLVKLNEKKLRAVDFLVKIGAVKSKNEARRVIQGGGLKINGEKVTDPNTEIEINGELKVKVGKKKFYRVVSG
2.- Obtenció dhomòlegs
2.1.- Homologia a nivell de seqüència
Sha realitzat un anàlisi dhomologies de seqüència de la tirosil-tRNA sintetasa dAquifex aeolicus a nivell de seqüència mitjançant un Blast a la base de dades Swissprot. Un cop analitzats els resultats obtinguts tenint en compte els E-values, les seqüències a partir de les quals es realitza, mitjançant el Clustalw, un alineament de seqüència són:
O67632 Aquifex aeolicus
P43836 Haemophilus influenzae
P56417 Helicobater pylori
P41256 Thiobacillus ferrooxidans
P75122 Mycoplasma pneumoniae
P22326 Bacillus subtilis
O83806 Treponema pallidum
O29482 Archaeoglobus fulgidus
P54577 Homo sapiens
P36421 Saccharomyces cerevisiae
O67115 Aquifex aeolicus
P00952 Bacillus stearothermophilus
Q99TD5 Staphilococcus aureus
Dels resultats obtinguts no sobserva cap gap o buit en cap regió present a totes les seqüències. No obstant, i tot i que els resultats obtinguts no són dolents, sha decidit abandonar aquest camí, ja que, la informació obtinguda mitjançant anàlisis estructurals serà més rellevant en el treball que ens ocupa. Decidits a abandonar, doncs, aquesta línia, tot el que segueixi a continuació farà referència a lanàlisi estructural de la proteïna, a partir de les homologies estructurals trobades al PDB.
1.2.- Homòlegs estructurals
Els homòlegs estructurals shan obtingut mitjançant un
Blast a PDB a partir de la seqüència O67632
en format FASTA de la tirosil-tRNA sintetasa dAquifex aeolicus. Els homòlegs
estructurals que sha escollit per modelar la proteïna són
els que es mostren a continuació:
|
Quadre 1. Homòlegs estructurals, en format pdb, de la proteïna problema.
1.2.1- Superposició destructures
Sha realitzat una superposició de les estructures homòlogues
per tal de determinar visualment les semblances i diferències de
les proteïnes homòlogues escollides. El resultat daquesta
superposició amb lStamp lhem visualitzat amb el Rasmol,
i evidencia lexistència de dos grups estructuralment diferents.
Un és format pels pdbs 1D2R i 1I6M corresponents a la tirosil-tRNA
sintetasa de Bacillus stearothermophilus, i laltre grup és
format duna banda pels pdbs 1JIL, 1JIK, 1JII, 1JIJ, corresponents a Staphilococcus
aureus, i daltra banda pels pdbs 1TYA, 1TYB i 1TYD, corresponents
a Bacillus stearothermophilus.
Fig 2. Superposició dels pdbs homòlegs a la proteïna problema. Es diferencien dos grups clarament diferenciats que corresponen a:
I: 1D2R i 1I6M
II: 1JIL, 1JIK, 1JII, 1JIJ,1TYA, 1TYB i 1TYD.
3.- Obtenció del perfil i alineament amb
HMM
Sha realitzat un perfil HMM amb cadascun dels dos grups dhomòlegs estructurals per tal desbrinar quin grup és més semblant a la tirosil-tRNA sintetasa dAquifex aeolicus i per tant més apte per modelar la proteïna problema. Els resultats de lhmmsearch, evidencien una major semblança entre la proteïna problema i els homòlegs estructurals del grup II, amb un score de 1.9e-20.
Donat que la tirosil-tRNA sintetasa dAquifex aeolicus presenta un grau de semblança molt més elevat amb els homòlegs estructurals del grup II, es modelarà la proteïna utilitzant aquest grup de templates, a partir de lalineament de la nostra proteïna problema amb el perfil de templates creats mitjançant lhmmalign. A aquest alineament se li retalla la cua C-terminal ja que no queda alineada i no aporta informació.
4.- Modelat de la proteïna amb Modeller
Mitjançant el programa Modeller sobté una primera
aproximació a lestructura tridimensional de la tirosil-tRNA sintetasa
dAquifex aeolicus. Aquest model sanalitza mitjançant el
Prosa
II i el Procheck, i són els que es mostren a continuació:
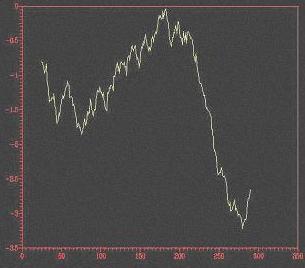
Fig. 3.: A: Resultat del primer modelatge. B:Espectre denergies del
Prosa II del model obtingut amb el Modeller per a la tirosil-tRNA sintetasa dAquifex aeolicus.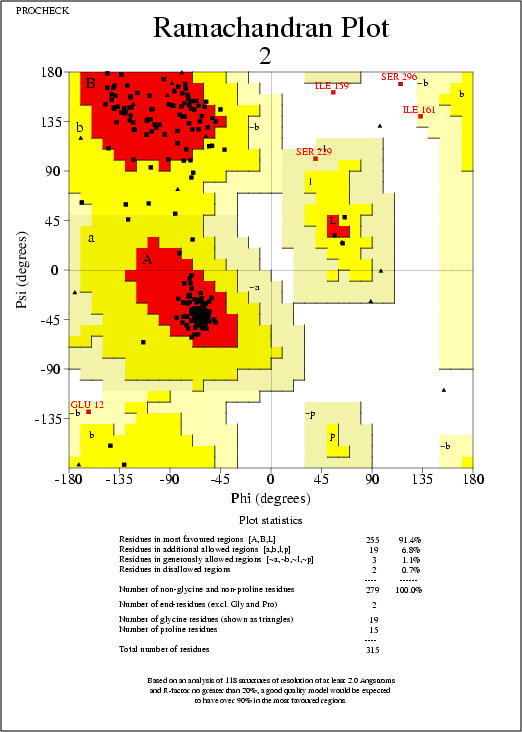 Fig. 4: Resultats del Procheck A: Resum. B: Mapa de Ramachandran
Fig. 4: Resultats del Procheck A: Resum. B: Mapa de Ramachandran
5- Optimització del model
5.1.- Modelatge dels loops
Observant lalineament obtingut a partir del perfil de HMM, saprecia un gap de 2 aminoàcids (IP) que no presenta homologia amb cap de la resta de proteïnes, i que es correspont a un loop estructural situat entre dues hèlix alfa. És en aquesta petita regió on sha centrat latenció per millorar el model mitjançant la cerca dun loop conegut que pugui omplir aquest petit buit. Shan trobat resultats per cobrir aquest petit gap, malgrat tot, els resultat obtinguts no han estat prou bons com per incorporar-los al model, ja que el valor dscore no ha estat suficientment bo.
Els resultats no són gaire bons, ja que el valor dels scores
és de 2 (aprox.) per als dos models obtinguts (per ser bons hauria
de ser < -50). Per tant, doncs, no ens serveix per optimitzar el model.
Abandonem aquesta via i es procedeix a la realització de la Dinàmica
Molecular.
5.2.- Minimització denergies amb Grumos
Sha realitzat una minimització denergies mitjançant Grumos i shan obtingut 10 models. El perfil energètic ha anat millorant durant loptimització, tal com es mostra en la figura 5B.
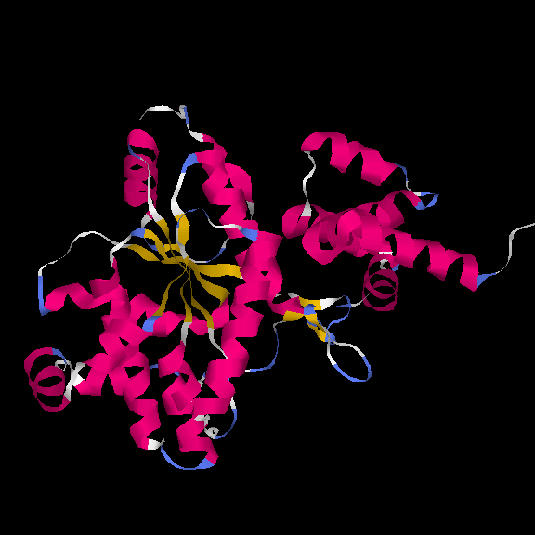
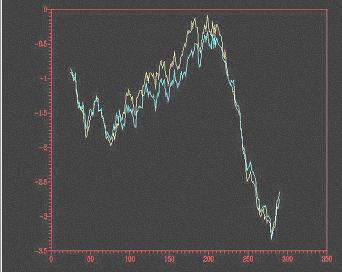
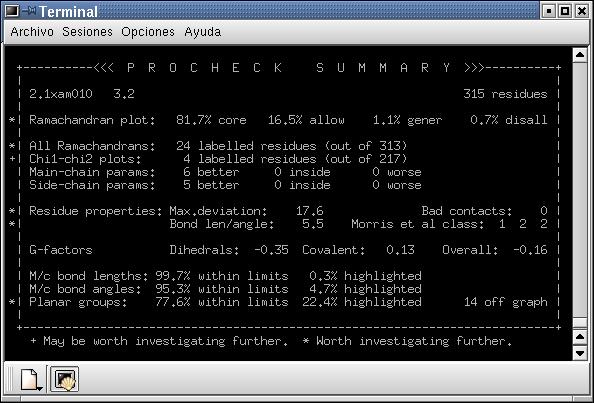

Fig. 5: A: Visualització del model optimitzat mitjançant el Rasmol.B: Comparació entre el primer i el darrer pas doptimització, obtinguda mitjançant el Prosa II. C: Resum del Procheck pel model optimitzat; D: Mapa de Ramachandran.
Tal com es pot observar en la figura 4, loptimització mitjançant
el Grumos ha permès millorar el model. En primer lloc sha
disminuït els nivells energètics, en segon lloc sha
reduït el nombre de contactes desfavorables de 16 a 1, i el nombre
de residus localitzats a la zona no permesa del mapa de Ramachandran ha
disminuït força. Malgrat tot, es veuen una sèrie de
punts, tot i que no són gaire, que estan a la zona dels no permesos.
Lanàlisi daquests residus revela la no rellevància funcional,
tot i que formen part de loops i no nhi ha cap que estigui formant part
duna hèlix alfa o làmina beta.
6.- Estudi de la Dinàmica Molecular
Després de minimitzar les energies mitjançant dinàmica molecular, no hi ha hagut variació significativa d'energies, tal com s'observa a la figura 6A i, comparant la figura 6B amb el sumari del Procheck obtingut en el la minimització d'energies amb el Grumos (figura 5C), no s'aprecien diferències significatives en els resultats. Això fa pensar que la minimització d'energies amb el Grumos ha sigut molt satisfactòria.
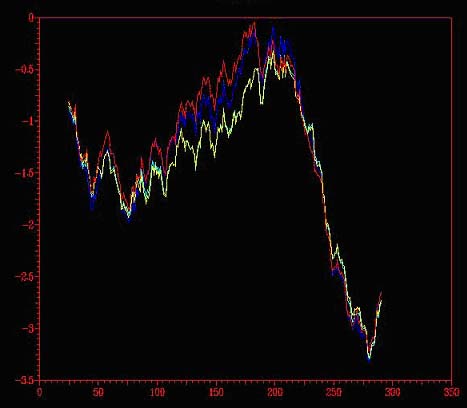
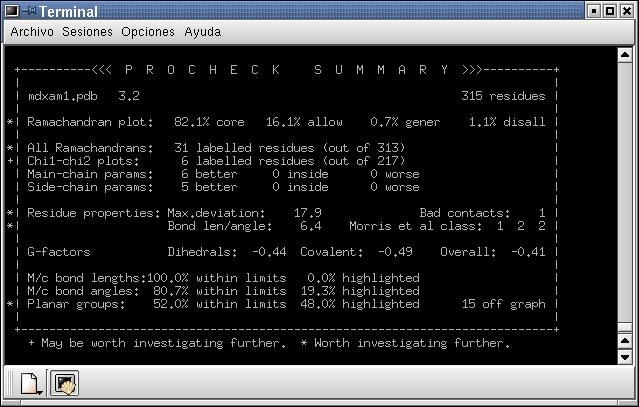

Fig. 6.: A: Comparació entre el model sense optimitzar (vermell), primer (blau fosc) i darrer pas doptimització (blau clar) i model optimització per dinàmica (groc), obtinguts mitjançant el Prosa II. B: Resum del Procheck pel model optimitzat per dinàmica molecular ; C: Mapa de Ramachandran.
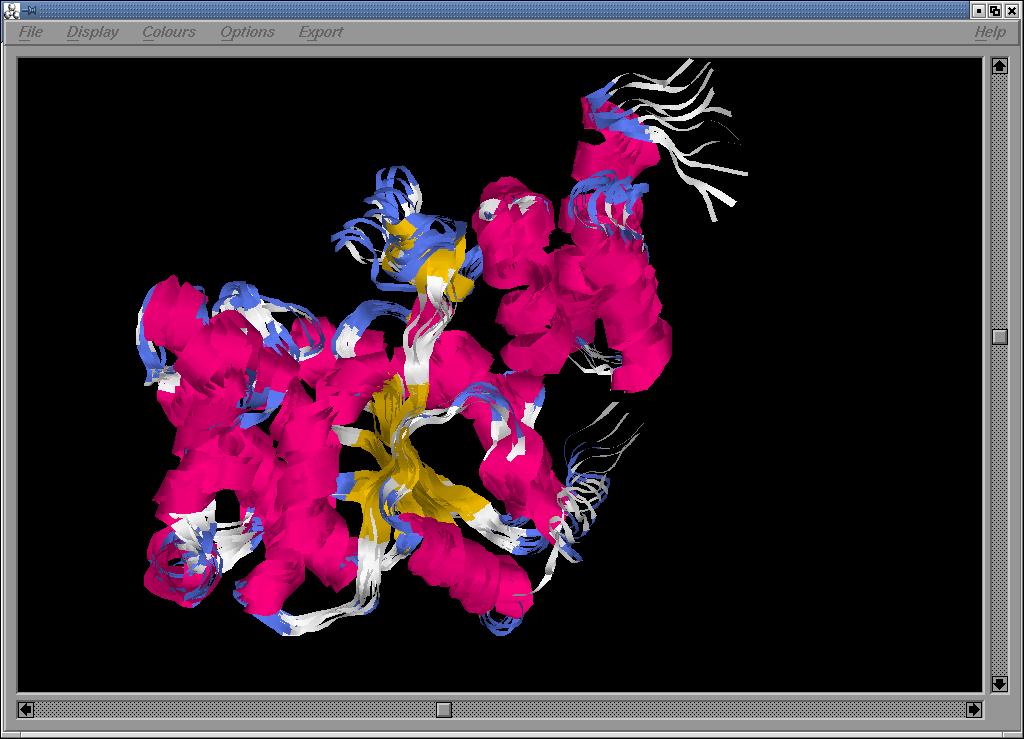
Lestudi de dinàmica de la proteïna permet analitzar la
flexibilitat de la proteïna. Una primera aproximació visual
del dinamisme de la proteïna es pot veure a continuació:
Fig. 7.: Diferents visions de la dinàmica molecular de la proteïna: cadena, estructura i ribbons, respectivament.
S'ha analitzat el dinamisme de la proteïna mitjançant
el B-factor. Aquí es mostren els resultats obtinguts:
Com s'observa a la figura, la regió corresponent a l'aminoàcid 225 és molt variable pel que fa a la seva flexibilitat. Tal com és d'esperar, aquesta regió correspon a un loop estructural.
Fig. 8.: Representació de les fluctuacions per a cada residu aminoacídic.
CONCLUSIONS
A partir de la seqüència en format FASTA de la tirosil-tRNA
sintetasa dAquifex aeolicus O67632 sha aconseguit modelar la proteïna
utilitzant 7 templates. Analitzant els resultats obtinguts sen
pot definir la seva estructura tridimensional.
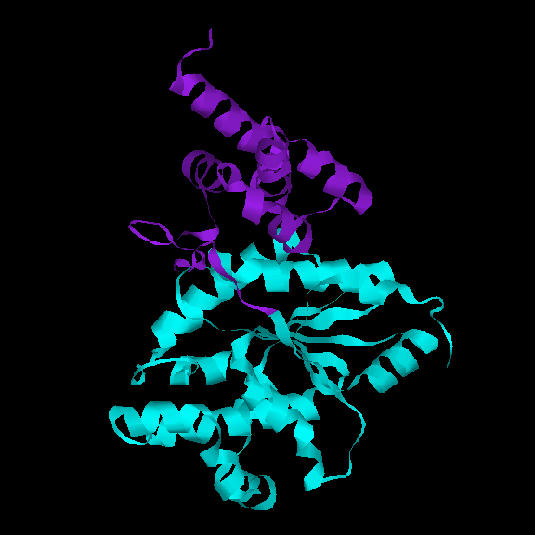
La tirosil-tRNA sintetasa d'Aquifex aeolicus és un enzim
dimèric. Cada subunitat consta de dos dominis: un domini catalític
(color turquesa i rosat a la figura 9), la funció del qual és
unir ATP i tirosina, i un domini format per alfa-hèlixs, de funció
desconeguda.
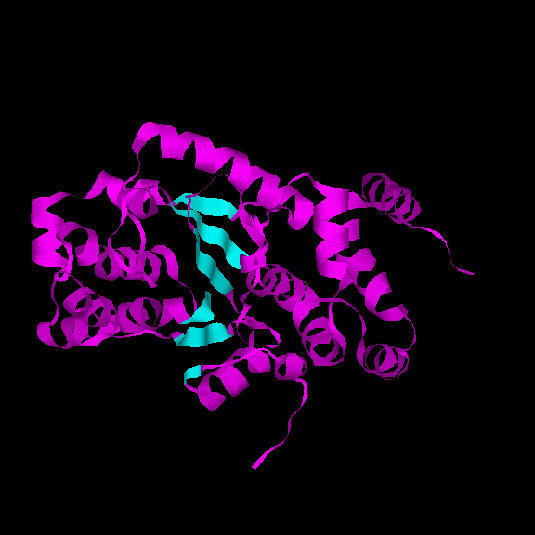
La regió central del domini catalític és de tipus alfa/beta (open beta-sheet). Consta d'una fulla beta de sis cadenes, la primera de les quals és antiparal.lela a la resta. El conjunt d'aquestes cadenes beta forma un nucli hidrofòbic envoltat per hèlixs alfa.Fig 9. Dominis de la tirosil-tRNA sintetasa d'Aquifex aeolicus. La part inferior (blau turquesa) correspon a un domini catalític amb plegament de Rossman. La part superior (lila) correspon a un segon domini format per hèlix alfa, amb funció desconeguda.
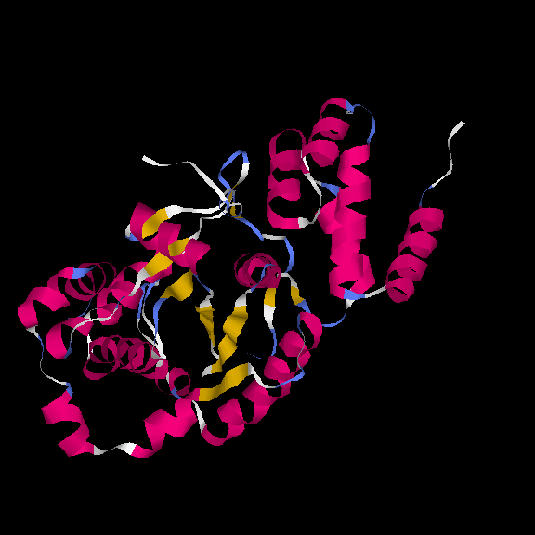
El centre actiu de l'enzim es troba en la fisura formada pels loops de l'extrem C-terminal de les cadenes beta. Aquesta fisura està formada pel canvi d'orientació que s'observa en el diagrama topològic. És en aquesta regió anomenada "topological switch point" on s'uneix l'ATP. Aquest tipus de plegament també es coneix amb el nom de Plegament de Rossman (Rossmanfold), el qual és característic d'unió a mononucleòtids (2).Fig. 10.: Domini catalític amb 6 làmines beta (marcades amb blau turquesa a la figura).
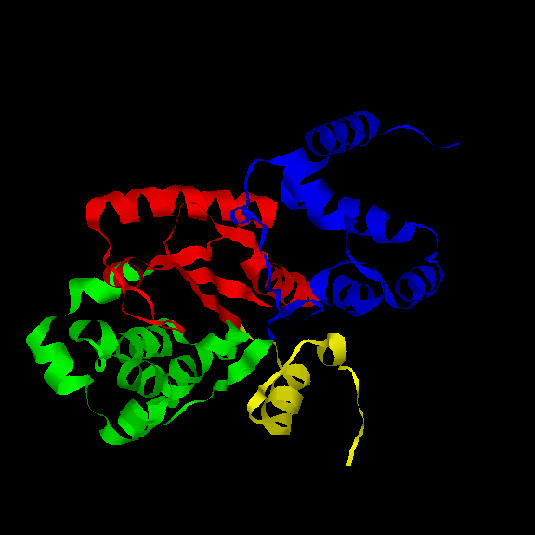
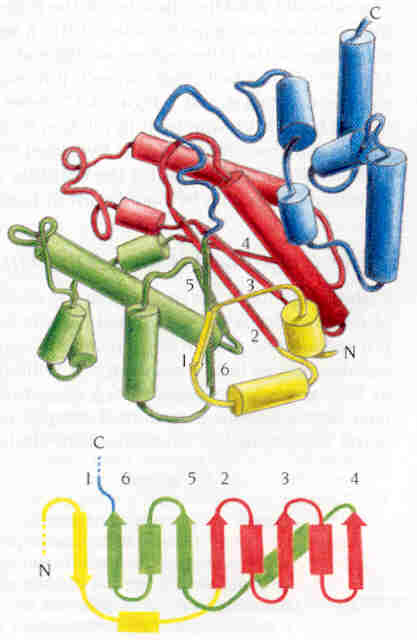
Fig. 11.: Diagrama esquemàtic de l'enzim tirosil-tRNA sintetasa d'Aquifex aeolicus. La regió central del domini catalític (vermell i verd) és una estructura alfa/beta de tipus open twisted amb 6 làmines beta paral.leles. El centre actiu està format pels extrems carboxi-terminals de les làmines beta 2 i 5, en la fisura generada pel canvi de sentit d'aquestes làmines beta. En groc, la primera làmina beta antiparal.lela a la resta.
Les hèlixs alfa que connecten les cadenes beta 2 i 3, i les cadenes beta 3 i 4, es troben situades a una banda de la làmina beta, mentre que les hèlixs alfa que connecten les cadenes beta 4 i 5 i les cadenes beta 5 i 6 es troben situades a l'altra banda de la làmina, tal com s'observa a la figura 11.
El centre actiu (groc a la figura 12) es localitza als extrems C-terminals
de les cadenes beta 2 i 5. El loop entre aquestes
cadenes beta i els seus alfa-hèlixs és el lloc d'unió
al substrat, el qual, concretament s'uneix a través dels residus
38-47 (loop 2) i 190-193 (loop 5). Al centre actiu s'hi uneix la tirosil-adenilat
mitjançant ponts d'hidrogen. Aquest substrat també s'uneix
als residus 173 i 177 de l'alfa hèlix que connecta les cadenes beta
4 i 5, i amb alguns residus de la cadena beta 2.
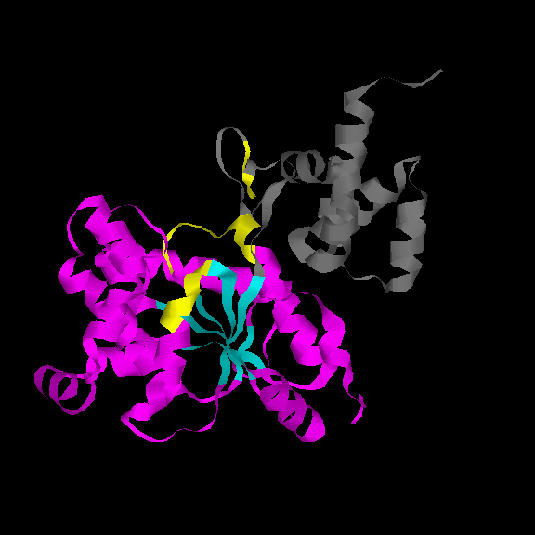
Fig. 12.: Centre actiu de la tirosil-tRNA sintetasa d'Aquifex aeolicus (en groc a la figura).
El coneixement de l'estructura i la funció de la tirosil-tRNA
sintetasa suposa un pas endavant en l'apassionant món d'aquesta
família de proteïnes, que van aparèixer en les primeres
etapes de l'origen de la vida. L'estudi de la seva estructura tridimensional
ens fa adonar de la importància que tenen en el manteniment de la
vida, ja que la seva "petjada" es troba en moltes altres proteïnes,
la qual cosa confirma el seu origen llunyà i ens aporta una peça
més en l'estudi del misteri de la vida.
1.- Arnez J., Moras D. Structural and functional considerations of the aminoacylation reaction. Trends in Biochemical Sciences 22: 211-216, 1997.BIBLIOGRAFIA
I si voleu saber més sobre les aminoacils... visiteu:
Bacardit M., Coll M., Gabernet N. Hostes
vingueren i a sorgir ens empenyeren! Origen endosimbiòtic dels
mitocondris
i arbre sense arrel de tots els organismes vius, a partir de les aminoacil
t-RNA sintetases.
Pràctiques devolució, 2001.
Bacardit M., Coll M., Gabernet N. Aminoacil-tRNA sintetases. http://www1.imim.es/courses/BioinformaticaUPF/projectes/1.6
M.Mercè Bacardit i Reguant, Montse Coll i Lladó, Núria Gabernet i Díaz i Jon Portuondo i Murguiondo
© Biologia Estructural-2002
Per qualsevol dubte
o aclariment no ho dubtis i escriu-nos un e-mail:
merce.bacardit01@campus.upf.es
montserrat.coll02@campus.upf.es
nuria.gabernet01@campus.upf.es
jon.portuondo01@campus.upf.es





{kind=link}
{kind=link}
{kind=link}