OBJECTIUS
1.-Fes servir XAM per a calcular el RMSD:
Calcula el millor RMSD entre les estructures de les hčlix 1,2,3 i 4 del directori de “practica_4/XAM”. Quins són els intervals de residus més adequats per tal d’obtenir el millor alineament estructural?
2.-Analitza la classificació de SCOP.
Fes una comparació estructural de 2 plegaments globin-like de la mateixa família de SCOP amb el programa XAM.
XAM
Xam es un programa de superposició s'estructures, que utilitzarem per a calcular el RMSD. La superposició que realitza és manual, hem d'introduir el rang d'amniŕcids a superposar.
El RMSD (root mean square deviation), es un terme que utilitzarem pe a mesurar a variabilitat entre les dos estructures comparades. La variabilitat pot ser mesurada en termes de distŕncia des de la mitjana. El RMSD es un promitg de la distancia a la mitjana.
En aquesta prŕctica calcularem el RMSD per a diverses estructures. Un valor de RMSD per sobre de 2A, ens indica que els estructures son considerablement variables. Si el valor estigués entre 3-5 A, encara podríen haber dubtes de si la superposició es bona, depenent de la complexitat de la proteďna. Un valor correcte hauria de ser menor a 1 A.
-Prŕctica:
Copiem el directori amb els arxius necessaris per a realitzar la prŕctica.
$cp -r /disc9/practica_4/XAM .
Executem XAM
$/disc9/Superposition/xam/xam
Dins d'aquest directori tenim l'estructura de 4 hčlixs:
-helix1.pdb
-helix2.pdb
-helix3.pdb
-helix4.pdb
La prŕctica consistirà en anar superposant diverses d'aquestes estructures, veure el RMSD y la observar superposició amb Rasmol.
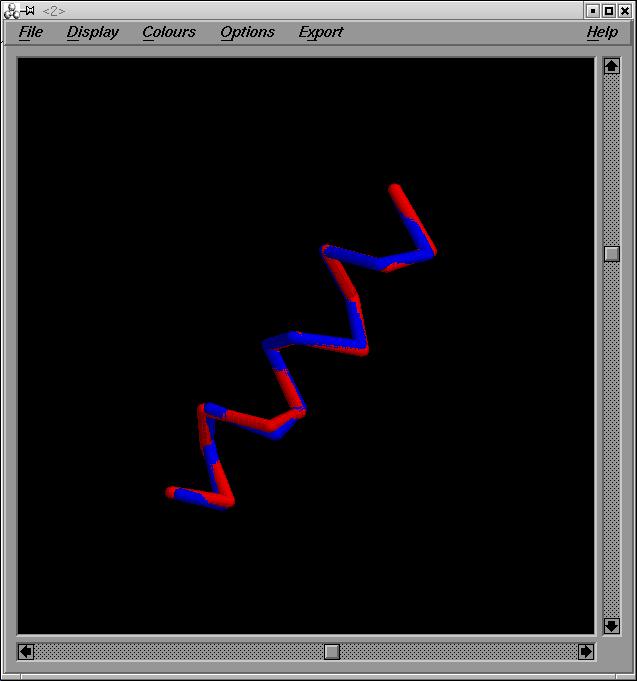
Hčlix 3 y Hčlix 4:
Primer de tot superposarem les estructures helix3.pdb i helix4.pdb. En aquest cas escollirem tots els residus (del 1 al 14). Obtenim com a output el fitxer helix_3_4.pdb i out3_4.
En el fitxer out3_4 podrem observar el valor de RMSD. En aquest cas es de 0'46 A.
Imatge superposició:helix_3_4
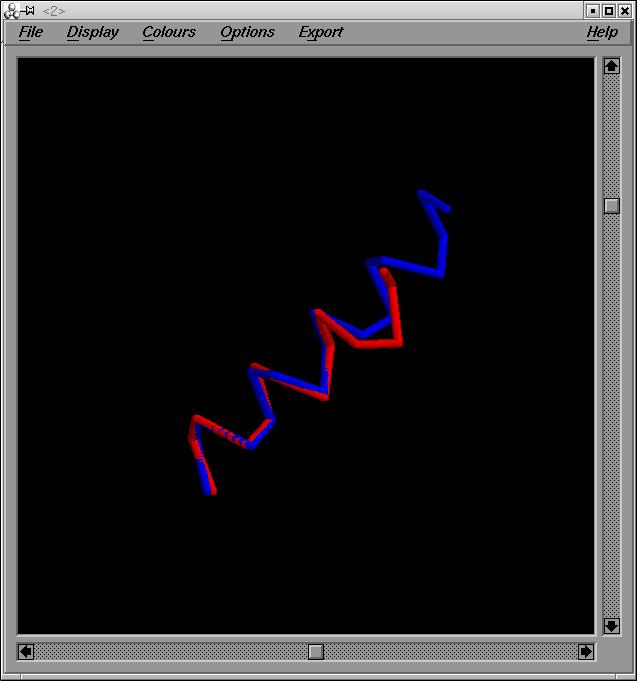
Helix 2 y Helix 3:
Superposarem les estructures helix2.pdb i helix4.pdb. En aquest cas ens trobem amb més problemes que amb l'anterior superposició. El primer problema amb el que ens trobem es que l’hčlix 2 te 18 aminoŕcids i l’hčlix 3 en te 14. Un altre problema és que l’hčlix 2 te una numeració diferent a la 3. Els de l'hčlix 2 van des del 37 al 54.
Per a solucionar aquests problemes haurem de superposar només fragments de les hčlixs. Escollirem fragments de 8 aminoŕcids. Superposem el fragment 37-44 de l'hčlix 2 i el fragment 1-8 de l'hčlix 8. hem escollit fragments de 8 aminoŕcids, perquècorresponen a dos voltes d'hčlix.
Obtenim els outputs helix_2_3.pdb i out2_3.
El valor de RMSD es de 0'29 A, per lo q podem assegurar que la superposició es bastant correcte.
Imatge superposició:helix_2_3
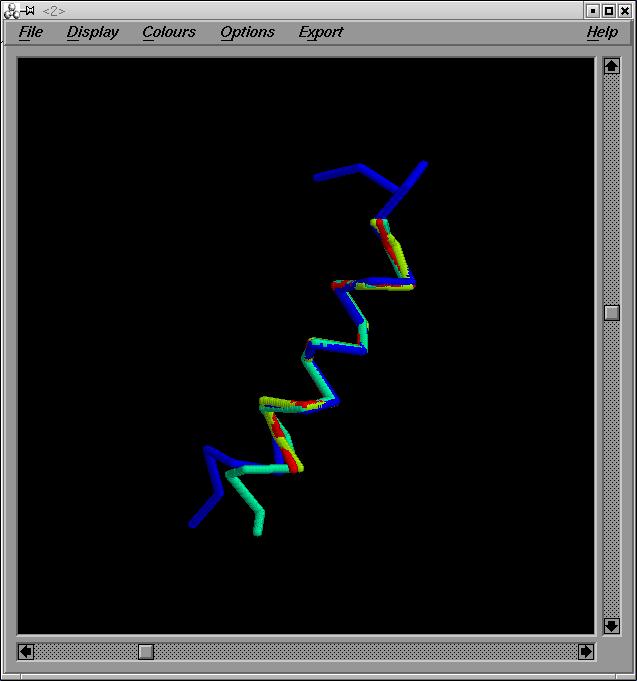
Hčlix 1, Hčlix 2, Hčlix 3, Hčlix 4:
Aquí ens trobarem amb els mateixos problemes que amb l'anterior superposició. En aquest cas a més tenim l'hčlix 1, que te una llargada de 23 aminoŕcids, i la seva numeració va del 1-23.
Hem escollit fragments de 13 aminoŕcids. De fet hem començat escollint fragments de 8 aminoŕcids, però he anat augmentant el tamany de l'alineament, ja que contra més residus alineem, més significatiu serà l'alineament:
-helix1 6-28
-helix2 37-49
-helix3 1-13
-helix4 1-13
helix_1_2_3_4.pdb i out1_2_3_4.
Els valors de RMSD obtinguts es bastant correcte. Oscilen entre 0'26 i 0'31.
Imatge superposició:helix_1_2_3_4
SCOP
Anirem a la web d'SCOP i escollirem dos proteďnes d ela mateixa família però de diferent espčcie, per a fer la superposició.
-Prŕctica:
Mirem a la web d'SCOP el nom de dos globines d'igual família però de diferent espčcies. Per això seguirem aquest camí: Tot Alfa-->Globin like-->Globins.
Un cop tenim escollides les proteďnes des de la web d'SCOP, baixarem la seva estructura des de la base de dades pdb local:
$/disc9/DB/pdb/pdb1a2m.ent.Z .
Descomprimim les estructures baixades:
$gunzip pdb1a2m.ent.Z
Les proteďnes escollides són:
1a6m :Sperm Whale (physeter catodon)
2mm1 :humŕ
Podem realitzar un alineament de les seqüčncies mitjançant Clustalw. Abans d'això haurem de realitzar un PDBtoSplitChain per tal d'obtenir les seqüčncies en format FASTA.
Alineament: globin.aln
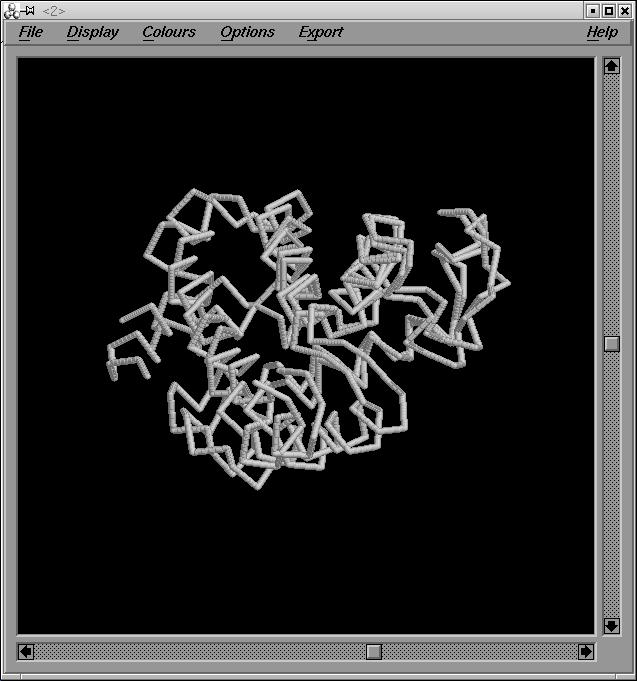
Realitzem el XAM de les dos globines. Podem veure el resultat en aquesta imatge:outglobins
{kind=link}
{kind=link}
{kind=link}
{kind=link}