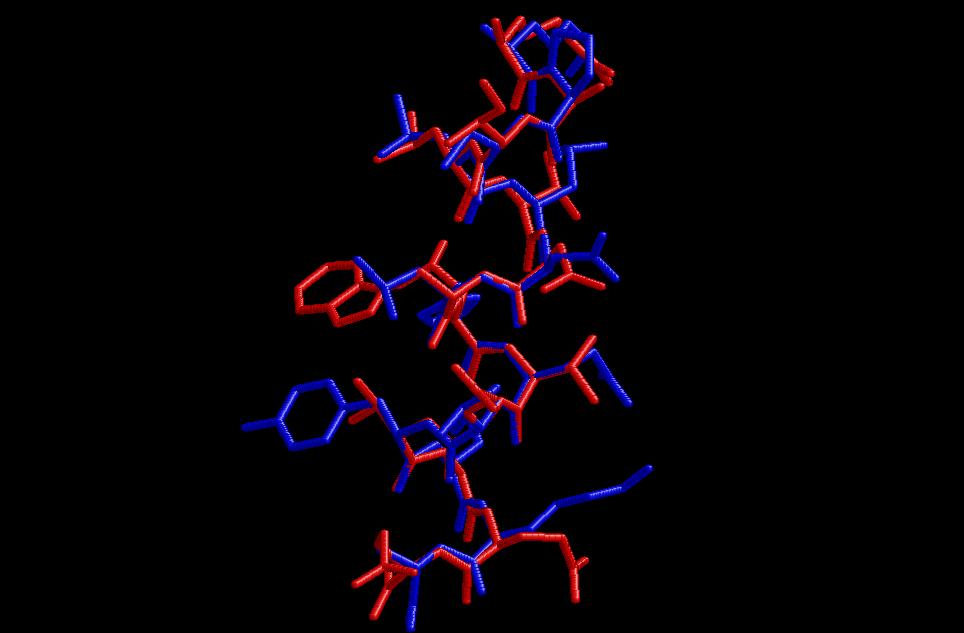
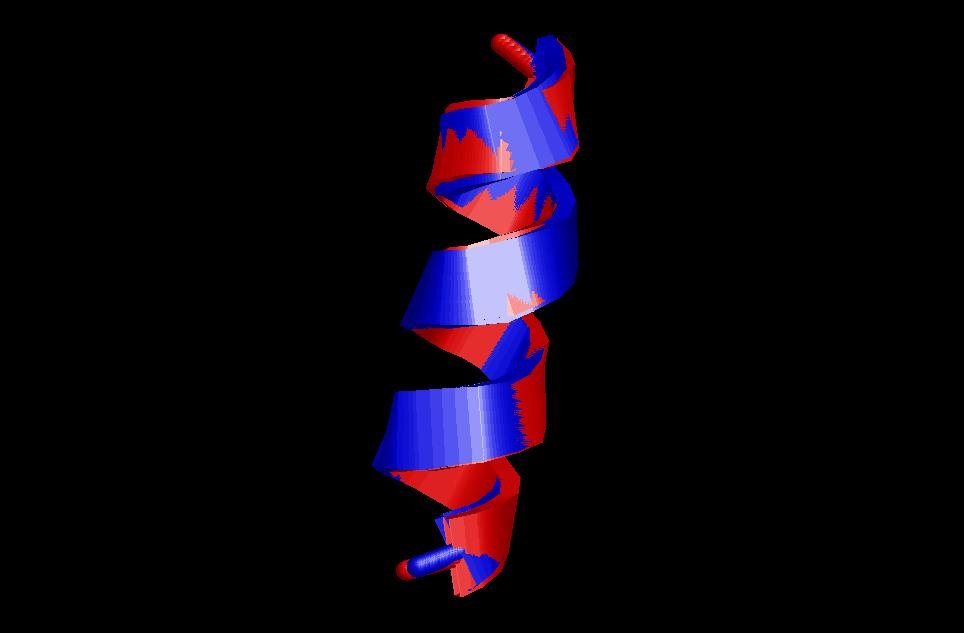
Objectiu: superposició d’estructures. Es tracta de buscar els valors de RMSD (Root Mean Square Deviation) més petits. Generalment els valors acceptables de RMSD estan entre 1 i 2, per sota de 2 pot ser aceptable i per sobre de 5 es considera molt dolent. Això en el cas de les proteïnes. Per les hèlix els valors han de ser molt més baixos, als voltants de 0,5 A)
1.
Anem a buscar les proteïnes que superposarem:
$ mkdir practica_4
$ cd practica_4
$ cp -r /disc9/practica_4/XAM .
$ cd XAM (en aquest directori hi ha 4 proteïnes que mirarem en el rasmol)
$ /disc9/bin/rasmol helix4.pdb (té 14 carbonis alfa)
2. I si l’obrim en un editor veurem el llistat dels
residus i les seves característiques
$ kwrite helix4.pdb
ATOM 1 N ARG 1 -9.673 209.345 81.811 ATOM 2 CA ARG 1 -10.776 208.517 81.308 ATOM 3 C ARG 1 -10.732 208.441 79.782 ATOM 4 O ARG 1 -10.902 207.368 79.205 ATOM 5 CB ARG 1 -12.123 209.035 81.829 ATOM 6 CG ARG 1 -13.326 208.138 81.552 ATOM 7 CD ARG 1 -14.610 208.986 81.525 ATOM 8 NE ARG 1 -15.793 208.374 80.889 ATOM 9 CZ ARG 1 -16.098 208.503 79.595 ATOM 10 NH1 ARG 1 -15.281 209.183 78.802 ATOM 11 NH2 ARG 1 -17.206 207.957 79.091 ATOM 12 N ALA 2 -10.476 209.592 79.159 ATOM 13 CA ALA 2 -10.322 209.753 77.707 ATOM 14 C ALA 2 -9.185 208.865 77.216 ATOM 15 O ALA 2 -9.315 208.110 76.264 ...
3. La comanda grep en ajuda a buscar un residu en concret:
$ grep CA helix4.pdb | more (en aquest cas busca els residus amb
carboni alfa)
ATOM 2 CA ARG 1 -10.776 208.517 81.308 ATOM 13 CA ALA 2 -10.322 209.753 77.707 ATOM 18 CA ILE 3 -6.946 208.110 77.497 ATOM 26 CA LEU 4 -8.694 205.023 78.799 ATOM 34 CA VAL 5 -11.632 204.919 76.373 ATOM 41 CA ASP 6 -9.258 205.640 73.481 ATOM 49 CA TRP 7 -7.138 202.576 74.472 ATOM 63 CA LEU 8 -10.328 200.516 74.574 ATOM 71 CA VAL 9 -11.030 201.504 70.957 ATOM 78 CA GLU 10 -7.584 200.169 70.066 ATOM 87 CA VAL 11 -8.041 196.955 72.054 ATOM 94 CA GLY 12 -11.304 196.522 70.174 ATOM 98 CA GLU 13 -9.627 196.947 66.770 ATOM 107 CA GLU 14 -6.766 194.607 67.704
4. Mirem el número de residus de cada una amb aquesta
mateixa comanda
$ grep CA helix3.pdb | more (també té 14 C alfa)
$ grep CA helix1.pdb | more (té 23 C alfa)
$ grep CA helix2.pdb | more (té 18 C alfa)
5. Superposició amb el programa XAM:
$ /disc9/Superposition/xam/xam
Fa una serie de preguntes com ara:
Output file name: output_3_4
Input file list? Or <cr>: (és per si tens moltes estructures i vols fer una llista de totes elles)
Structure 1 or <cr>: helix3.pdb
Structure 2 or <cr>: helix4.pdb
All molecules are the same type if OK <cr>: (és important especificar quins residus vols superposar en el cas que les proteïnes no tinguin el mateix número de residus; ara no és el cas)
Option: 1 (càlcul de l’RMSD; pot fer moltes altres coses com el càlcul de la superfície (7=surface_no_atmo o 8=surface_with_atoms), càlcul de ponts d’hidrogen, etc.)
First and last residue of frag 1: 1 14 (en el cas que vulgués superposar fragments ho hauria d’indicar aquí)
Option: 1
Output file for superimposed structure? Or <cr>: helix_3_4.pdb
Output fmt: 4 (bPDB: això fa que ho pugui veure en rasmol després; la opció PDB crea un fitxer amb la suma dels residus de les dues molècules sense diferenciar les dues cadenes)
First and last residues of frag 1: 1 14
Option: 0 (per sortir)
6. Obrim el resultat amb un editor:
$ emacs output_3_4
Observem un quadre d’aquest estil on hi ha els valors de RMSD:# RMSD table # # 1 2 # 1 0.46 # 2 0.00
En aquest case ens ha donat un RMSD de 0,46 A, que és for�a bo. Això ens diu que la superposició és bona; de fet, no ens sorprèn perquè les molècules són gairebé idèntiques. Els problemes arribaran quan s'intenti superposar estructures més complicades amb plegaments alfa i beta i proteïnes més grans.
7. Mirem la superposició amb rasmol:
$ /disc9/bin/rasmol helix_3_4.pdb (és interessant mirar la opció STICKS per veure que com de diferents són les cadenes laterals)
Aquí tenim la superposició en backbone i en cartoons. Amb la segona es pot pensar que la superposició és quasi perfecte, per&oagrave; he adjuntat l'altre perquè es vegi que les cadenes laterals no tenen cap mena de coincidència. Això ja era d'esperar.
8. A continuació superposarem les hèlix 2 i 3
que tenen el problema afegit de no estar formada pel mateix
nombre d’aa. La hèlix 2 té 54 residus dels
quals 18 són carbonis alfa: del 37 al 54. La hèlix
3 ja hem vist abans que en tenia 14.
$ /disc9/Superposition/xam/xam
Hem de variar lleugerament la resposta d’algunes preguntes que ens fa el programa:
Fragments for superposition...first & last residue of frag1: 41 54 (correspont a la hèlix 2)
Fragments for superposition...first & last residue of frag1: 1 14 (correspon a la hèlix 3)
Option: 1 (RMSD)
Output: helix_2_3.pdb
Option: 4 (bPDB)
Fragments...: 41 54
Fragments...: 1 14
Option: 0 (sortir)
9. Visualitzem els resultants:
$ emacs output_2_3 (tenim un valor de RMSD de 0,67 A, el qual és lleugerament major que en el cas anterior però encara està dins del rang acceptable; és també una bona superposició)
# RMSD table # # 1 2 # 1 0.67 # 2 0.00$ /disc9/bin/rasmol helix_2_3.pdb
Aquí es pot veure com una de les hèlix és menor que l'altre, però com que nosaltres hem escollit els fragments a superposar, així ens ho ha fet. Si no haguéssim especificar els residus, no haurí pogut fer el XAM perquè necessita sembpre superposar fragments de la mateixa longitud. Altres opcions del programa, ja més complicades, só la de superposar diferents fragments de la mateixa molècula.
1. Descripció de cada hèlix:
Hèlix 1: 1-23 (23aa)
Hèlix 2: 37-54 (18aa)
Hèlix 3: 1-14 (14aa)
Hèlix 4: 1-14 (14aa)
2. Fem un primer intent superposant les hèlix 3 i 4
completes, els 14 primers residus de l’hèlix 1 i els
residus 37-50 de la hèlix 2. El resultat ens dóna
els següents valors d’RMSD:
La primera linia, que correspon a la superposició de la hèlix 1 amb la 2, la 3 i la 4 és la que dóna uns valors d’RMSD massa alts. Com s'ha explicat al principi de la pràctica, el rang d'acceptació està entre 0,5 i 1 (i depenen de si la molècula és molt gran, pot arribar fins a 2). De totes maneres, aquests valors es poden millorar, superposant altres fragments de les molècules. Tanmanteix els resultats de la superposició de la hèlix 2 amb la 3 i la 4 i la mateixa 3 amb la 4, ja són prou bons. De manera que els fragments utilitzats en aquestes hèlix els mantindrem, mentre que buscarem altres combinacions per la hèlix 1.# RMSD table # # 1 2 3 4 # 1 2.97 3.10 3.08 # 2 0.00 0.54 0.42 # 3 0.00 0.00 0.46 # 4 0.00 0.00 0.00 #
3. Provem altres combinacions: variem els aa que afagem de la hèlix 1, ara superposarem el fragment del residu 6 al 19 (això no s’escull a l’atzar, ho deduïm de veure el resultat del xam en el rasmol i observar quins punts són els de més coincidència i quins els de menys). Si mirem el resultat del rasmol; que aquí no adjunto es pot comprovar quina és la zona que menys ve s'ha superposat i que per tant ha generat un RMSD tant alt. Així intuim d'on eliminem els residus que sobren i per on. Les altres hèlix segueixen igual. Resultat:
# RMSD table # # 1 2 3 4 # 1 0.60 0.70 0.69 # 2 0.00 0.54 0.42 # 3 0.00 0.00 0.46 # 4 0.00 0.00 0.00
4. Ha millorat bastant però intentarem ajustar encara
una mica més els valors de RMSD. Com ja he dit abans aquests ajusts s'aconsegueixen anar provant la superposició de diferents fragments i amb una mica d'intuició. Ho aconseguim finalment
després de varis intents amb la superposició dels
següents fragments de cada hèlix:
Hèlix 1: 7-20
Hèlix 2: 37-50
Hèlix 3: 1-14
Hèlix 4: 1-14
El resultat és el següent:
# RMSD table # # 1 2 3 4 # 1 0.72 0.65 0.50 # 2 0.00 0.54 0.42 # 3 0.00 0.00 0.46 # 4 0.00 0.00 0.00
Si ho mirem amb rasmol tenim una idea tridimensional de l'alineament:
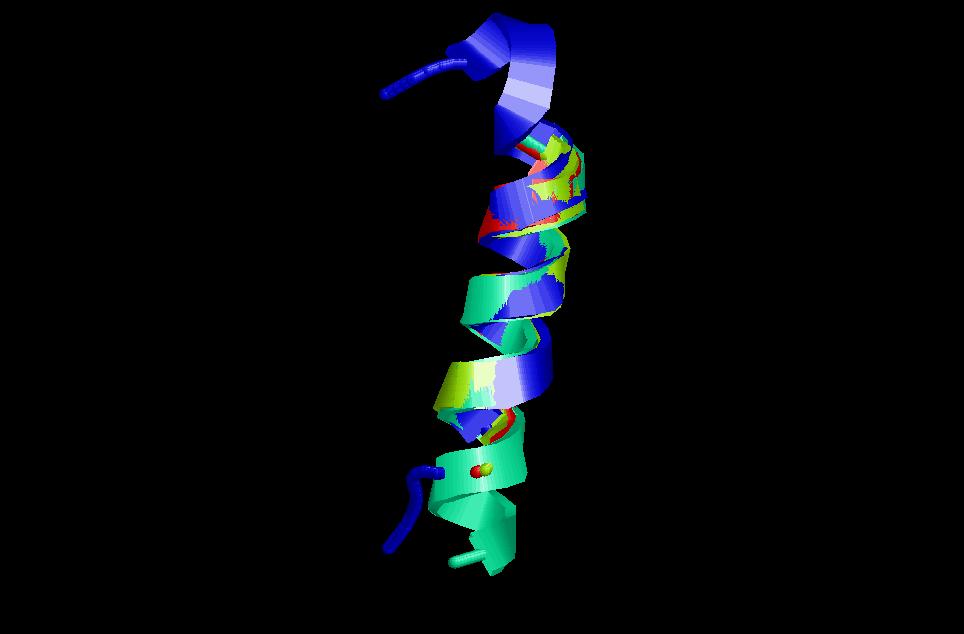


Conclusió: Aquestes tres imatges ens donen una idea tridimensional de l'alineament però tampoc ens podem fiar nomé dels resultats visuals. La primera imatge està generada amb cartoons, la segona amb strands i la tercera amb backbone, totes elles opcions del rasmol. I ràpidament es veu com la hèlix de color verd té un petit fragment terminal que no se superposa amb les altres. En els altres intents intentàvem superposar aquesta zona i per això obteníem valors de RSMD tant alts. Una altra raó la tenim en la hèlix de color blua, la qual té un gir diferent en un del extrems, que no està en cap de les altres hèlix i que per això hem hagut de deixar fora de l'alineament. La lectura que se'n pot fer de tot plegat és que la superposició d'estructures és prou complicada. Si un exemple tant simple com aquestm ens ha fet provar 3 o 4 combinacions diferents per arribar a l'alineament més acurat, quan es tracti d'una molècula més gran, amb hèlix alfa i làmines beta, el treball serà increïble.