Objectius: Alineament de dues proteïnes estructuralment similars però seqüncialment molt diferents. El mètode STAMP es basa en el càlcul de distàncies entre el carboni alfa d’un residu i els seus veïns en les dues proteïnes que volem comparar. Es fa un paràmetre que depengui de la diferència de distàncies: ř = |di1-dj1| + |di2-dj2| +... Si els entorns són similars aquestes diferències seran petites.
STAMP: es tracta d'un programa d'alineament de seqüències proteiques basat en l'estructura tridimensional d'aquestes. Tot i que els outputs siguin fitxers de seqüències alineades, aquest programa és d'estructures 3D. Per tant, l'STAMP és un programa que no es pot utilitzar si s'està treballant amb proteïnes d'estructura desconeguda. A més a més, el programa et permet fer una superposició molt acurada: canvi de la direcció d'strands, selecció de un determinat fragment de cada proteïna,... entre d'altres opcions. Per realitzar aquesta pràctica fem servir una opció més directa perquè no necessitem cap d'aquestes opcions.
El programa està basat en l'equació de Argos & Rossmann, que expressa la probabilitat de l'equivalència estructural de residus. STAMP utilitza l'algorisme Smith Waterman, que determina el millor camí d'alineament a través d'una matriu de puntuació.
1. Ja no treballarem amb bash:
$ tsch
] source /disc9/cshrc
] cd practica_4
] mkdir STAMP
] cd STAMP
] cp /disc9/practica_4/STAMP/EXAMPLES/globin.tar .
] tar xvf globin.tar (descomprimim l’arxiu amb el que realitzarem la pràctica)
] cd globin (conté 6 globines diferents)
2. Creem un fitxer on hi haurà les proteïnes que superposarem:
] kwrite globin.domains
Hi escrivim el següent:
./1ecd.pdb 1ecd {ALL}
./2hhba.pdb 2hhba {chain A}
./2hhbb.pdb 2hhbb {chain B}
./1lh1.pdb 1lh1 {ALL}
./2lhb.pdb 2lhb {ALL}
./4mbn.pdb 4mbn {ALL}
ALL si es superposarà tot el que hi ha al pdb (tota la proteïna) i chain per especificar la cadena (per això és necessari fer un PDBtoSplitChain previament).
3. Executem el programa STAMP:
] stamp -l globin.domains -rough -n 2 -prefix globin
El resultat és una serie de fitxers: globin.1, globin.2, globin.3, globin.4 i globin.5, que corresponen respectivament a l’alineament de les dues seqüències més semblants, d’aquest alineament amb la que més s’hi assembla, etc.
4. El que ens interessa és l’últim: globin.5, perquè serà on hi tindrà les 6 globines:
] kwrite globin.5
LLLLLL ?????? 1 0.47035 1.88040 6.85700 TSSSST ?????? 1 0.51135 1.98110 7.43700 EAAEPP ?????? 1 0.57037 1.87782 8.27200 SADGAE ?????? 1 0.62733 1.77785 9.07800 QEQEDE ?????? 1 0.72035 1.61182 10.39400 AKIWKK ?????? 1 0.76933 1.49735 11.08700 ATSQTS ?????? 1 0.77336 1.48066 11.14400 LKTLNA ?????? 1 0.82835 1.32079 11.92200 VIVVVV ?????? 1 0.87034 1.18472 12.51600 KRQLKT ?????? 1 0.84736 1.24954 12.19100 SSAHAA ?????? 1 0.82036 1.29586 11.80900 SASVAL ?????? 1 0.85634 1.16145 12.31800 WWFWWW ?????? 1 0.85238 1.16222 12.26200 EADAGG ?????? 1 0.79534 1.31527 11.45500 EPKKK ????? FVVVVK ?????? 1 0.66635 1.48884 9.63000 NY EGV ?? ??? AS AA ?? ?? NTKDHN ?????? 1 0.50032 1.72566 7.28100 IY VAV ?? ??? PEGAGD ?????? 1 0.33832 2.17075 4.98900 KT GEE ?? ??? HSDHYV ?????? 1 0.67632 1.36284 9.77100 TGPGGG ?????? 1 0.73237 1.53619 10.56400 HVVQAG ?????? 1 0.68035 1.62140 9.82800 ...
5. Transformem aquest alineament vertical a un d'horitzontal, que sabrem llegir millor:
] aconvertMod2.pl -in b -out c < globin5 | more
...i per pantalla veiem:
CLUSTAL W(1.60) multiple sequence alignment 1hl1 --------GALTESQAALVKSSWEEFNANIPKHTHRFFILVLEIAPAAKDLFSFLKGTSE 2lhb PIVDTGSVAPLSAAEKTKIRSAWAPVYSTYETSGVDILVKFFTSTPAAQEFFPKFKGLTT 1ecd ----------LSADQISTVQASFDKV--K-G-DPVGILYAVFKADPSIMAKFTQFAG-KD 4mbn ---------VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRFKHLKT 2hhba ---------VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-D--- 2hhbb --------VHLTPEEKSAVTALWG-KV-NVDEVGGEALGRLLVVYPWTQRFFESFGDLST space ------------------------------------------------------------ 1hl1_dssp --------???????????????????????????????????????????????????? 2lhb_dssp ???????????????????????????????????????????????????????????? 1ecd_dssp ----------????????????????--?-?-?????????????????????????-?? 4mbn_dssp ---------??????????????????????????????????????????????????? 2hhba_dssp ---------??????????????????????????????????????????????-?--- 2hhbb_dssp --------????????????????-??-???????????????????????????????? ...
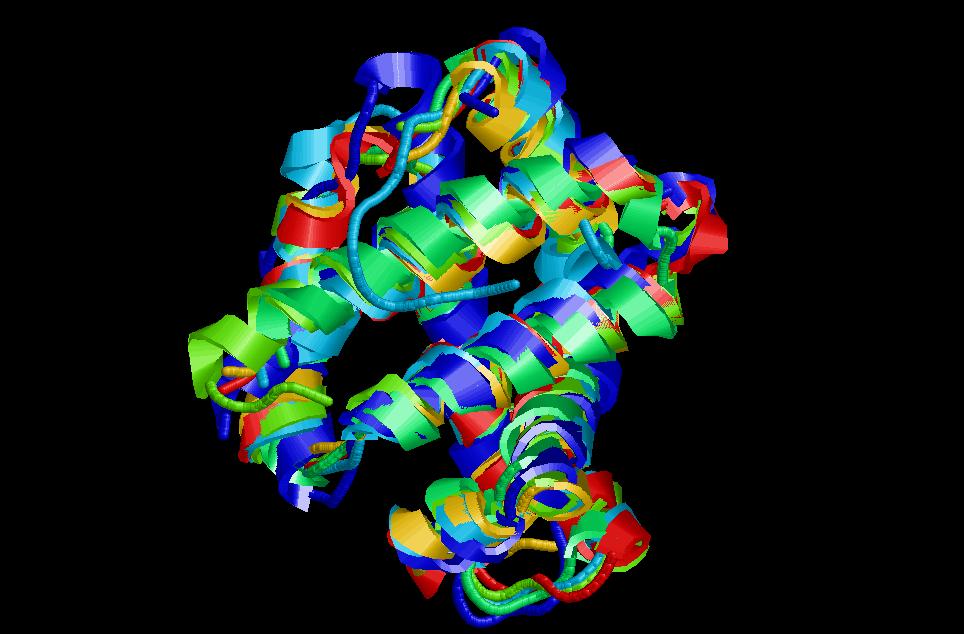
6. Només cal mirar el pdb final amb el rasmol i ja tenim el resultat:
] rasmol globin.5.pdb
Conclusions: és una bona superposició. Es tracta de proteïnes amb plegament domini tot alfa i és fàcilment reconeixible en aquesta forografia. Les hèlix són les regions més conservades i és on sobserva una superposició més clara.