Pràctica 5.4
Objectius: volem comprovar que l'alineament sigui
correcte i verificar que l'estructura tingui sentit químic
i estereoquímicament semblant. El programa que ens ho
permet fer és el procheck
PROCHECK: Aquest programa verifica la qualitat
estereoquímica de l'estructura de la proteïna,
produint un gran nombre de PostScripts (.ps) i un fitxer
sumari (.sum), que contenen l'anàlisis
geomètric en conjunt i residu a residu. L'explicació detallada de cada fitxer de sortida està explicada en el desenvolupament de la pràctica. Per fer
córrer el programa necessitem una sèrie de
requisits:
procheck filename [chain] resolution
filename = el fitxer de coordenades del model
[chain] = opcional (no l'usem nosaltres, aquesta
opció)
resolution = nombre real, és la resolució
més alta de totes les proteïna templates que
utilitzem
Desenvolupament de la pràctica:
1. Farem córrer aquest programa pels quatre models que
tenim: P11018.B99990001 i P11018.B99990002 per clustalw i per
HM
$ procheck-single P11018.B99990001 2.2
2. Els resultats del fitxer sumary (P11018.sum) dels
quatre són els següents:
Primer el dos de Clustalw:
P11018.B99990001. Tenim 18 bad contacts que serien àtoms amb impediments estèrics i que per tant generen "contactes irregulars" o desfavorables pel conjunt energètic de la molècula. Els altres valors que hem d'interpretar són els de Ramachandran plot. El primer percentatge és el número de residus que es troben en zones molt bones o permeses. El segon correspon a zones no tant favorables com les altres per&oagrave; encara permeses (allow). El tercer (gener) és el percentatge de residus que es troben en una zona poc favorable però permesa "generosament". I finalment els classificats com disall són aquells que definitivament estan en una zona no permeta del mapa de Ramachandran. Tenen angles completament forçats i desfavorables. Per tant, uns bons resultats series: els dos primers valors molt alts, en contraposició a valors baixos de gener i disall; i la minimització del nombre de bad contacts.
+----------<<< P R O C H E C K S U M M A R Y >>>----------+
| |
| P11018.B99990001 2.2 319 residues |
| |
*| Ramachandran plot: 86.3% core 11.4% allow 1.1% gener 1.1% disall |
| |
*| All Ramachandrans: 11 labelled residues (out of 317) |
+| Chi1-chi2 plots: 3 labelled residues (out of 180) |
| Main-chain params: 6 better 0 inside 0 worse |
| Side-chain params: 5 better 0 inside 0 worse |
| |
*| Residue properties: Max.deviation: 16.9 Bad contacts: 18 |
*| Bond len/angle: 7.4 Morris et al class: 1 1 2 |
+| 1 cis-peptides |
| G-factors Dihedrals: -0.12 Covalent: -0.27 Overall: -0.17 |
| |
| M/c bond lengths: 99.2% within limits 0.8% highlighted |
*| M/c bond angles: 91.7% within limits 8.3% highlighted 1 off graph |
| Planar groups: 100.0% within limits 0.0% highlighted |
| |
+----------------------------------------------------------------------------+
+ May be worth investigating further. * Worth investigating further.
P11018.B99990002: En aquest, tenim menys bad contacts, la qual cosa 6eacute,s millor. Però el percentatge d'àtoms en zones prohibides del mapa de Ramachandran 6eacute;s pitjor i també ha disminuit aquells que es troben en zones m6eacute,s permeses. Per tant, en conclusió, aquest model seria lleugerament pitjor que l'anterior.
+----------<<< P R O C H E C K S U M M A R Y >>>----------+
| |
| P11018.B99990002 2.2 319 residues |
| |
*| Ramachandran plot: 81.9% core 15.9% allow 0.7% gener 1.5% disall |
| |
*| All Ramachandrans: 16 labelled residues (out of 317) |
+| Chi1-chi2 plots: 1 labelled residues (out of 180) |
| Main-chain params: 6 better 0 inside 0 worse |
| Side-chain params: 5 better 0 inside 0 worse |
| |
*| Residue properties: Max.deviation: 7.1 Bad contacts: 11 |
*| Bond len/angle: 10.5 Morris et al class: 1 1 2 |
+| 1 cis-peptides |
| G-factors Dihedrals: -0.11 Covalent: -0.37 Overall: -0.20 |
| |
| M/c bond lengths: 98.3% within limits 1.7% highlighted |
*| M/c bond angles: 91.4% within limits 8.6% highlighted 3 off graph |
| Planar groups: 100.0% within limits 0.0% highlighted |
| |
+----------------------------------------------------------------------------+
+ May be worth investigating further. * Worth investigating further.
I ara els de HMM:
P11018.B99990001: No són uns resultats molt bons perquè el percentatge de gener &ecaute;s massa alt, tot i que es compensa amb el baix que trobem com a disall.
+----------<<< P R O C H E C K S U M M A R Y >>>----------+
| |
| P11018.B99990001 2.2 319 residues |
| |
*| Ramachandran plot: 83.0% core 14.0% allow 2.2% gener 0.7% disall |
| |
*| All Ramachandrans: 16 labelled residues (out of 317) |
*| Chi1-chi2 plots: 7 labelled residues (out of 180) |
| Main-chain params: 6 better 0 inside 0 worse |
| Side-chain params: 5 better 0 inside 0 worse |
| |
*| Residue properties: Max.deviation: 7.5 Bad contacts: 17 |
*| Bond len/angle: 11.1 Morris et al class: 1 1 2 |
+| 1 cis-peptides |
+| G-factors Dihedrals: -0.16 Covalent: -0.52 Overall: -0.28 |
| |
| M/c bond lengths: 97.1% within limits 2.9% highlighted |
*| M/c bond angles: 89.0% within limits 11.0% highlighted 7 off graph |
| Planar groups: 100.0% within limits 0.0% highlighted |
| |
+----------------------------------------------------------------------------+
+ May be worth investigating further. * Worth investigating further.
P11018.B99990002: Potser, aquest seria el pitjor del 4 perquè és el que té més bad contacts i presenta uns percentatges altíssims de disall i gener. De fet, aquesta evaluació global dels 4 models és bastant relativa perquè estem analitzant molts termes diferents i mentres un model pot ser bo per alguns d'ells no ho és pels altres. Per això també ens interessa mirar els altres outputs del procheck que són gràfics postscript que ens aportaran més informació.
+----------<<< P R O C H E C K S U M M A R Y >>>----------+
| |
| P11018.B99990002 2.2 319 residues |
| |
*| Ramachandran plot: 81.5% core 14.4% allow 2.6% gener 1.5% disall |
| |
*| All Ramachandrans: 20 labelled residues (out of 317) |
+| Chi1-chi2 plots: 4 labelled residues (out of 180) |
| Main-chain params: 6 better 0 inside 0 worse |
| Side-chain params: 5 better 0 inside 0 worse |
| |
*| Residue properties: Max.deviation: 9.5 Bad contacts: 19 |
*| Bond len/angle: 17.4 Morris et al class: 1 1 2 |
+| 1 cis-peptides |
+| G-factors Dihedrals: -0.21 Covalent: -0.82 Overall: -0.42 |
| |
*| M/c bond lengths: 96.2% within limits 3.8% highlighted 3 off graph |
*| M/c bond angles: 89.0% within limits 11.0% highlighted 14 off graph |
| Planar groups: 100.0% within limits 0.0% highlighted |
| |
+----------------------------------------------------------------------------+
+ May be worth investigating further. * Worth investigating further.
3. I a continuació analitzem un per un els
postscripts perquè cada un ens aporta una
informació diferent, tot i que el més important és el mapa de Ramachandrann:
$ ghostview P11018_01.ps
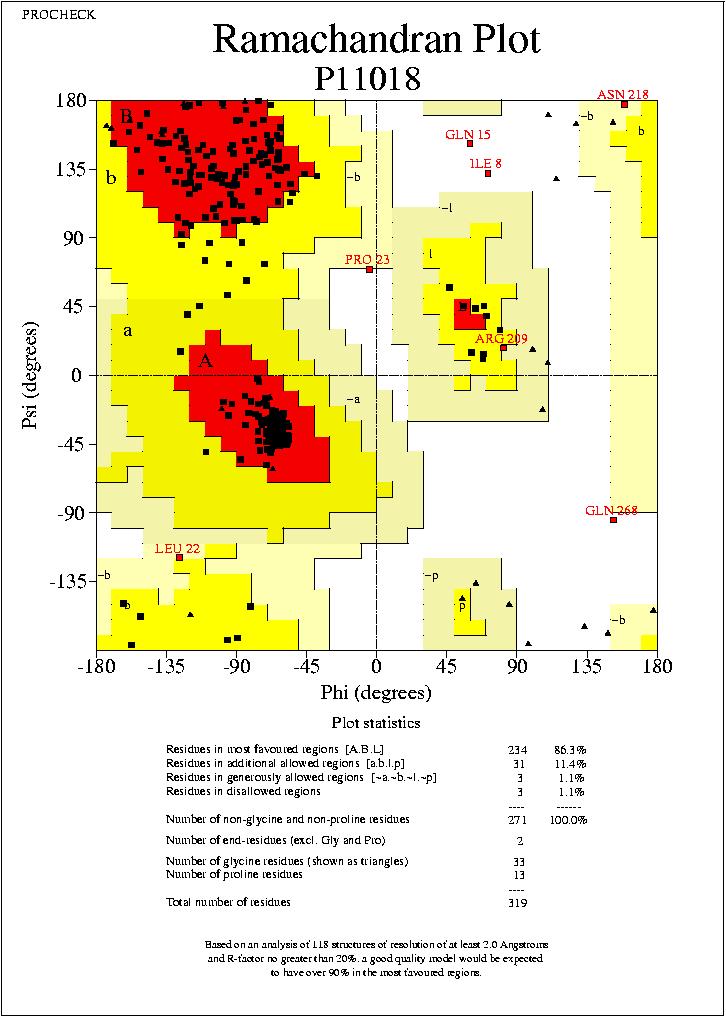 Mapa de Ramachandran: ens ensenya la torsió dels
angles phi-psi per tots els residus en l'estructura. Els residus
de glicina estan identificats per separat per triangles. Els
colors i les sombres en el mapa representen diferents regions,
descrites per Morris et al. (1992): l'àrea més fosc
(color vermell) correspon a les regions del core que
represeneten la combinació més favorable de valors
de phi-psi. Idealment, hom voldria tenir més del 90% de
residus en aquestes regions del core. El percentatge de
residus en la refgió del core és un dels
millors indicatius de la qualitat de l'estereoquímica.
Mapa de Ramachandran: ens ensenya la torsió dels
angles phi-psi per tots els residus en l'estructura. Els residus
de glicina estan identificats per separat per triangles. Els
colors i les sombres en el mapa representen diferents regions,
descrites per Morris et al. (1992): l'àrea més fosc
(color vermell) correspon a les regions del core que
represeneten la combinació més favorable de valors
de phi-psi. Idealment, hom voldria tenir més del 90% de
residus en aquestes regions del core. El percentatge de
residus en la refgió del core és un dels
millors indicatius de la qualitat de l'estereoquímica.
$ ghostview P11018_02.ps
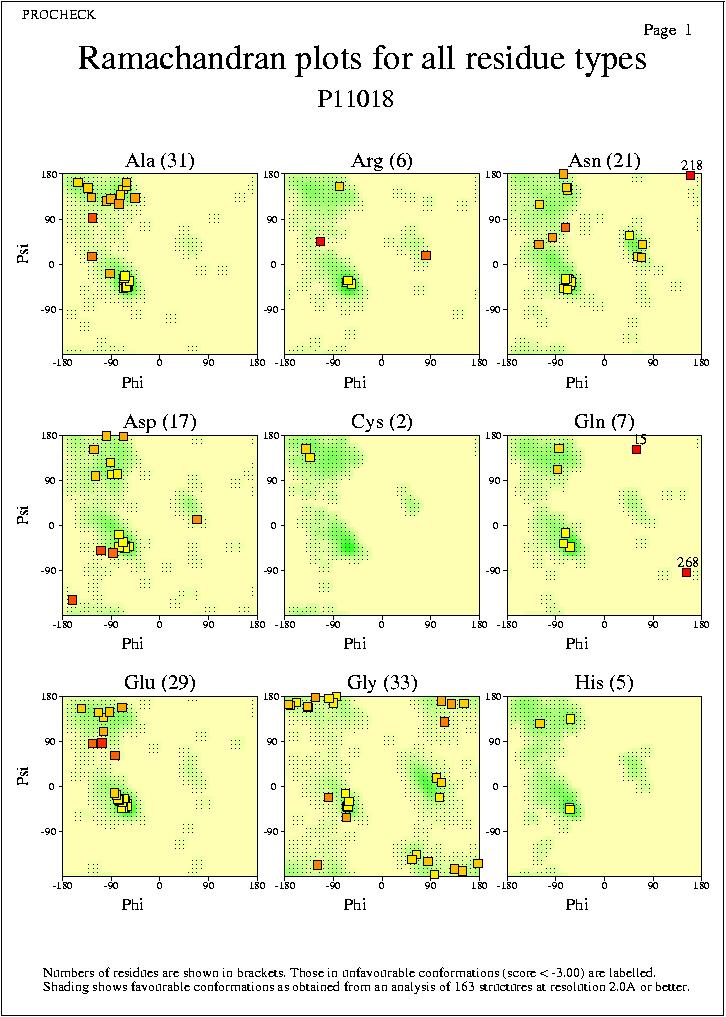 Mapa de Ramachandran per tots els tipus de residu: ens
mostra mapes de Ramacandran separats, un per cada un dels 20
tipus d'aminoàcids diferents. Com més fosca sigui
l'àrea sombrejada de cada mapa, més favorable
serà la regió. El criteri amb el qual s'han
establert aques sombres ve de l'anàlisi de 163 cadenes
proteiques no homòlogues i d'alta resolució,
escollides de estructures cristal·litzades i analitzades
per raigs X a una resolució de 2.0 A (o millor, amb un
R-factor no més gan del 20%). Els números entre
parèntesis, al costat del nom de cada residu, indiquen el
nombre total de punts que conté el mapa. El número
vermell per sota d'aquest és el número de residus
d'aquell aa que hi ha en la proteïna.
Mapa de Ramachandran per tots els tipus de residu: ens
mostra mapes de Ramacandran separats, un per cada un dels 20
tipus d'aminoàcids diferents. Com més fosca sigui
l'àrea sombrejada de cada mapa, més favorable
serà la regió. El criteri amb el qual s'han
establert aques sombres ve de l'anàlisi de 163 cadenes
proteiques no homòlogues i d'alta resolució,
escollides de estructures cristal·litzades i analitzades
per raigs X a una resolució de 2.0 A (o millor, amb un
R-factor no més gan del 20%). Els números entre
parèntesis, al costat del nom de cada residu, indiquen el
nombre total de punts que conté el mapa. El número
vermell per sota d'aquest és el número de residus
d'aquell aa que hi ha en la proteïna.
$ ghostview P11018_03.ps
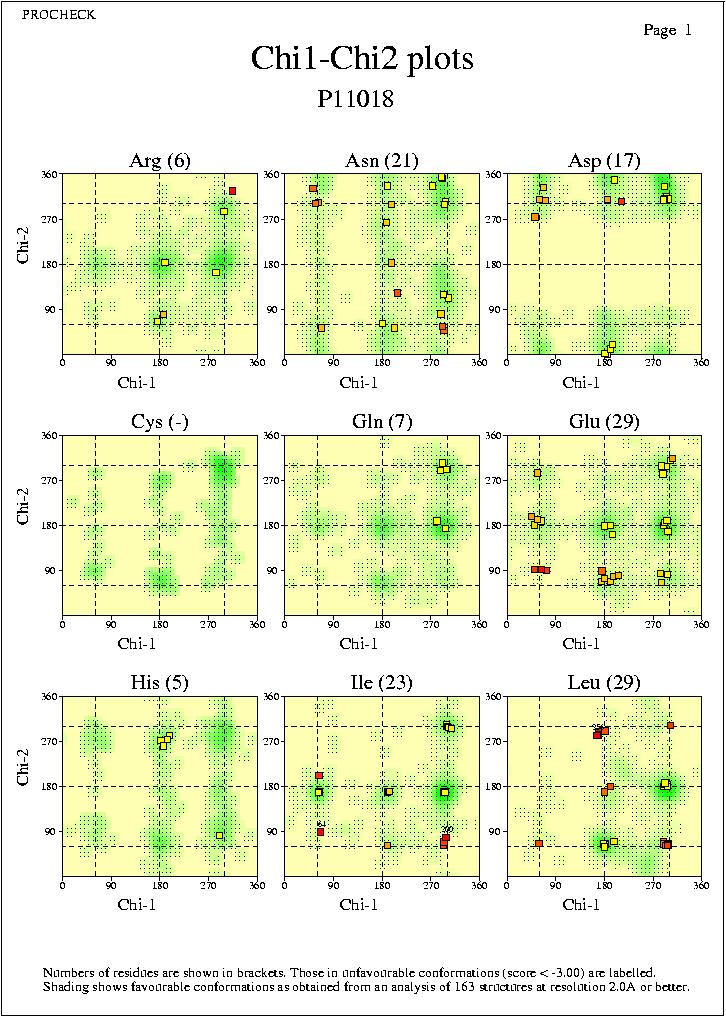 Chi1-chi2 plots: És la combinació de l'angle
de torsió chi1-chi2 de la cadena lateral de tots els tipus
de residus que la tinguin suficienment llarga com per tenir
ambdós angles chi. El sombrejat en cada mapa indica com
n'és de favorable cada regió; la part més
fosca és la més favorable. El criteri que determina
aquestes zones es va generar tal i com està indicat en
l'explicació dels mapes de Ramachandran per tots els
tipus de residus
Chi1-chi2 plots: És la combinació de l'angle
de torsió chi1-chi2 de la cadena lateral de tots els tipus
de residus que la tinguin suficienment llarga com per tenir
ambdós angles chi. El sombrejat en cada mapa indica com
n'és de favorable cada regió; la part més
fosca és la més favorable. El criteri que determina
aquestes zones es va generar tal i com està indicat en
l'explicació dels mapes de Ramachandran per tots els
tipus de residus
$ ghostview P11018_04.ps
Paràmetres de la cadena principal: Els sis
gràfics que conté aquest postscript ens mostren com
és l'estructura (representada per un quadradet negre) en
comparació amb estructures ben refinades a la mateixa
resolució. La banda lila de cada gràfic és
el resultat de les estructures ben refinades; la linia central
que la separa en dos és la mitjana, mentre que la resta de
banda lila correspon a la variació de la desviació
estàndar respecte la mitja. Com més tinguem el
nostre model (quadradet negre) dins la franja, millor, i si
està sobre la franja central, perfecte. Entendre amb
exactitud cada un dels gràfics és complicat
però breument resumeixo que hi podem trobar en cada un
d'ells:
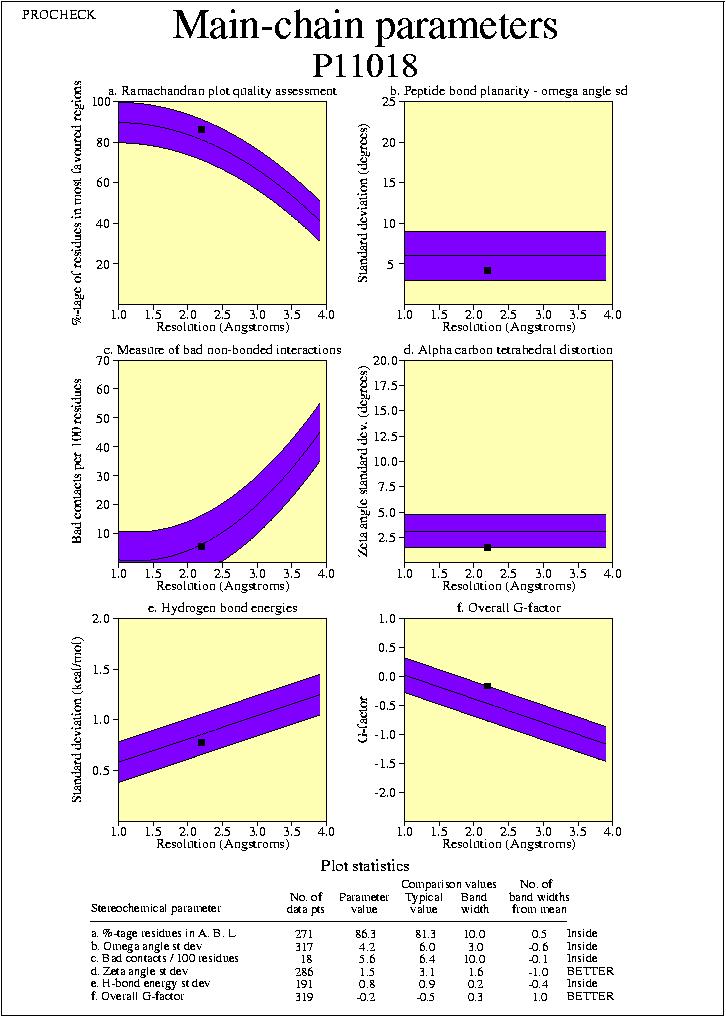
1.Qualitat del mapa de Ramachandran.
2.Planaritat del enllaços peptídics.
3.Interaccions no enllaçants incorrectes.
4.Distorsió del Calfa tetrahèdril.
5.Energia dels ponts d'hidrogen a la cadena principal.
6.G-factor.
$ ghostview P11018_05.ps
Paràmetres de les cadenes laterals: correspon al
que hem explicat en el postscript anterior però per les
cadenes laterals dels residus.
$ ghostview P11018_06.ps
Propietats dels residus: Els gràfics i diagrames
ens ensenyen com varien les propietats geomètriques dels
residus al llarg de la seqüpencia. Això ens
dóna un idea visual de quines regions semblen ser
consistenment més pobres o amb geometria poc usual (potser
perquè estan poc definides) i quines tenen una geometria
més normal.
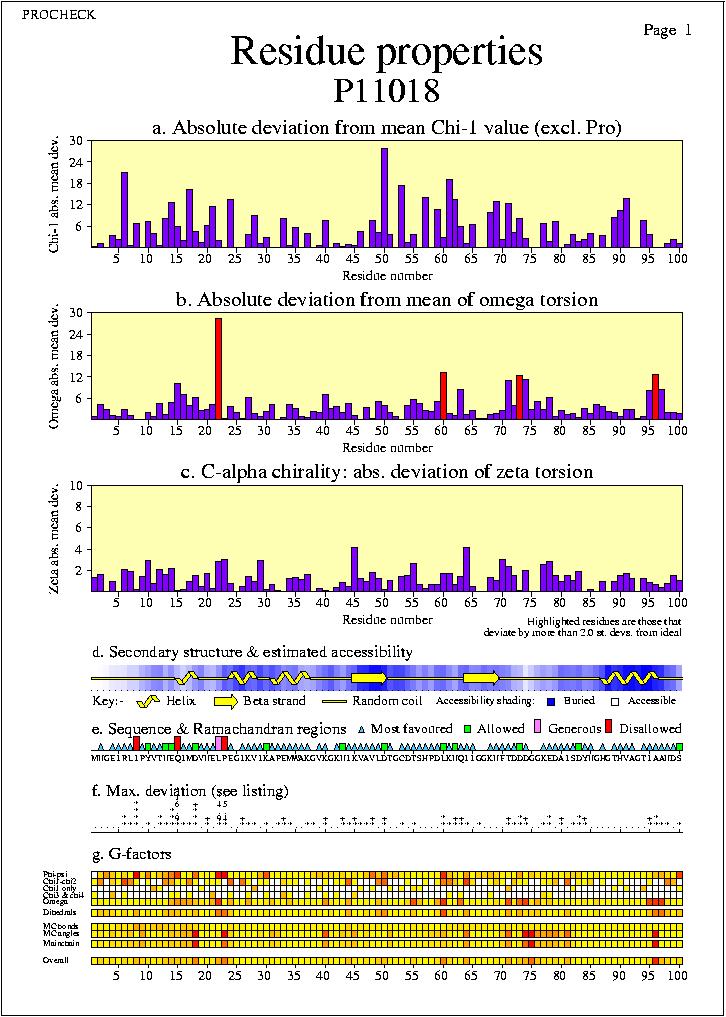
$ ghostview P11018_07.ps
Distribució de la llargada dels enllaços en la
cadena principal:L'histograma mostra la distribució de
cada una de les diferents llargades dels enllaços de la
cadena principal en l'estructura. La linia central continua
correspon al valor de la mitjana per petites molècules,
mentre les linies discontinues a ambdós cantons
corresponen a la desviació estàndar per
molècules petites també.
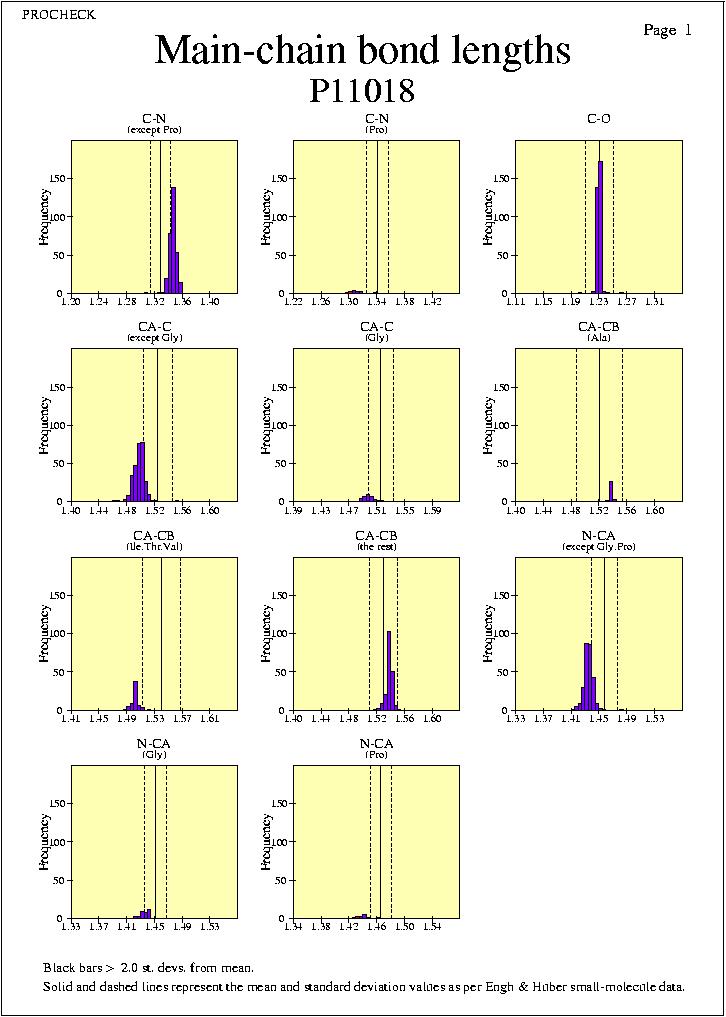
$ ghostview P11018_08.ps
Distribució dels angles d'enllaços en la cadena
principal: l'explicació de cada línia dels
histogrames és la mateixa que hi ha en l'apartat
anterior.
$ ghostview P11018_09.ps
Distàncies RMS de planaritat: Aquests histogrames
mosrten l'RMSD pels diferents grups planars en l'estructura. Les
línies discontinues indiquen diferents valors ideals per
anells aromàtics (Phe, Tyr, Trp i His) i per grups
terminals planars (Arg, Asn, Asp, Gln i Glu). Els valors per
defecte són 0.03A i 0.02A, respectivament.
$ ghostview P11018_10.ps
Mapes geomètrics distorsionats: Correspon a una
sèrie de gràfics que mostren la distorsió
d'altres mapes que ja hem explicat en altres postscripts (angles
enllaçants de la cadena principal, grups planars,...entre
d'alrtes).
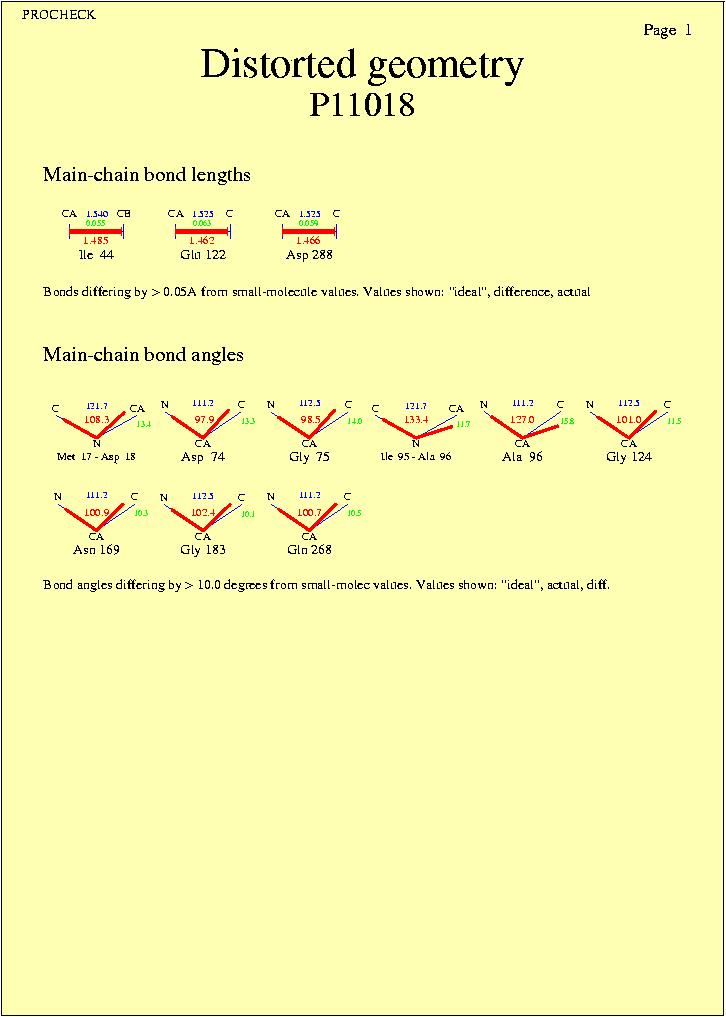
4. Un cop hem analitzat mínimament el resultat del
procheck, fem un càlcul predictiu de l'estructura
secundària del model:
$ dssp P11018.B99990001 model_01_C.dssp
(com que això s'ha de fer també pels 4 models, en
nom de l'output ha de ser indicatiu de a quin model es refereix;
en aquest cas seria model acabat en 01 del ClustalW, per Hiden
Markov el símbol que faré servir és H)
$ emacs model_01_C.dssp
Per pantalla ens apareix un fitxer ple de números i valors indesxifrables.
5. Ho passem a format .pir:
$ aliss.pl model_01_C.dssp > model_01_C.pir
$ emacs model.pir
>P1;model_01_C.dsspSeq
model_01_C.dsspSeq
MNGEIRLIPYVTNEQIMDVNELPEGIKVIKAPEMWAKGVKGKNIKVAVLDTGCDTSHPDLKNQIIGGKNFTDDDGGKEDA
ISDYNGHGTHVAGTIAANDSNGGIAGVAPEASLLIVKVLGGENGSGQYEWIINGINYAVEQKVDIISMSLGGPSDVPELK
EAVKNAVKNGVLVVCAAGNEGDGDERTEELSYPAAYNEVIAVGSVSVARELSEFSNANKEIDLVAPGENILSTLPNKKYG
KLTGTSMAAPHVSGALALIKSYEEESFQRKLSESEVFAQLIRRTLPLDIAKTLAGNGFLYLTAPDELAEKAEQSHLLTL*
>P1;model_01_C.dsspSS
model_01_C.dsspSS
-------------SHHHHTTTS-HHHHHTTHHHHHHHT---TT-EEEEEES---TT-TTSTTTEEEEEE-S-SSSS---T
T--SSSHHHHHHHHHH---SSSS---SSTT-EEEEEE-S-SS-SS--HHHHHHHHHHHHHHT-SEEEE---BS---HHHH
HHHHHHHHTT-EEEEE--S-----TT-----BTTTSTTSEEEEEE-TTS-B-TTS--STT--EEEE-SSEEEEETTTEEE
EE-SHHHHHHHHHHHHHHHHHHS--STTTT--HHHHHHHHHHT-B--SS-HHHHTT-B--SHHHHS-S---S-------*
I aquí ja tinc seqüència i l'estructura
secundària predita (on H: hèlix alfa, S: girs en un
loop, i E: fulla beta)
6. Ho convertime en format clustalW:
$ aconvertMod2.pl -in p -out c < model_01_C.pir >
model_01_C.clw
$ emacs model_01_C.clw
CLUSTAL W(1.60) multiple sequence alignment
model.dsspSeq MNGEIRLIPYVTNEQIMDVNELPEGIKVIKAPEMWAKGVKGKNIKVAVLDTGCDTSHPDL
model.dsspSS -------------SHHHHTTTS-HHHHHTTHHHHHHHT---TT-EEEEEES---TT-TTS
model.dsspSeq KNQIIGGKNFTDDDGGKEDAISDYNGHGTHVAGTIAANDSNGGIAGVAPEASLLIVKVLG
model.dsspSS TTTEEEEEE-S-SSSS---TT--SSSHHHHHHHHHH---SSSS---SSTT-EEEEEE-S-
model.dsspSeq GENGSGQYEWIINGINYAVEQKVDIISMSLGGPSDVPELKEAVKNAVKNGVLVVCAAGNE
model.dsspSS SS-SS--HHHHHHHHHHHHHHT-SEEEE---BS---HHHHHHHHHHHHTT-EEEEE--S-
model.dsspSeq GDGDERTEELSYPAAYNEVIAVGSVSVARELSEFSNANKEIDLVAPGENILSTLPNKKYG
model.dsspSS ----TT-----BTTTSTTSEEEEEE-TTS-B-TTS--STT--EEEE-SSEEEEETTTEEE
model.dsspSeq KLTGTSMAAPHVSGALALIKSYEEESFQRKLSESEVFAQLIRRTLPLDIAKTLAGNGFLY
model.dsspSS EE-SHHHHHHHHHHHHHHHHHHS--STTTT--HHHHHHHHHHT-B--SS-HHHHTT-B--
model.dsspSeq LTAPDELAEKAEQSHLLTL
model.dsspSS SHHHHS-S---S-------
Nota: Només adjunto el resultat de fer-ho amb el model de clustal 01 perquè més endavant (a la pràctica 6.2 quan haguem triat dos dels models ho tornarem a fer per comparar-los entre ells, i crec que aquell resultat és molt més interessant.
ÍNDEX