Pràctica 7
Objectius: Ja tenim el model escollit, i hem
comprovat que fos una bona elecció. L'últim pas
serà la optimització geomètrica d'aquest. Es
tracta de minimitzar l'energia potencial de sistema tant com
sigui possible. Ho farem a partir del programa grumos. Aquest
programa té tres passos bàsics:
1. Càlcul de
la topologia de la molècula
2. Minimització
energètica
3. Dinàmica molecular.
Desenvolupament de la pràctica:
1. Primerament s'ha de tenir en compte que és un
programa difícil d'executar, té moltes opcions i
has d'introduir molt paràmetres. Abans de començar
hem de complir una sèrie de requisits:
A. Hem d'assegurar-nos que quan hem tallat el model
pels extrems per millorar els gràfics del prosa, ho hem
fet correctament. El fitxer pdb del model ha d'acabar amb les
lletres TER o END i les coordenades de
l'últim àtom han de ser les de OXT. Per tant haurem
de fer uns petits canvis en el pdb. En comptes d'eliminar tots
els àtoms de tots els residus que sobreven, deixem el
primer àtom (N) de l'últim residu que eliminem.
Això només ho hem de fer en l'extrem C terminal.
És a dir, si havíem de tallar 9 residus per aquell
extrem, i la nostra proteïna és de 322
aminoàcids, eliminem els àtoms dels residus 322-315
complets, del residu 314 eliminem tots els àtoms excepte
el que està just per sota del primer àtom del
residu 313. Representa que les coordenades d'aquell àtom
coincideixen amb les de l'OXT si la proteïna, de forma
natural, s'acabés per allà. Alehores els canvis
hauran de ser: el nom del residu, el número i afegir-hi
TER en la darrera línia del pdb.
B. El segon requisit que ens exigeix el programa és que
identifiquem els ponts disulfur del nostre model. Per això
obrim el pdb amb el rasmol, seleccionem les cys i fem que calculi
la distància entre elles. Segons sigui aquesta
diatància major o menor decidirem si s'hi pot formar o no
un pont dissulfur. Si fos així hem de saber exactament els
residus que són les cys perquè el GRUMOS ens
demanarà que les identifique.
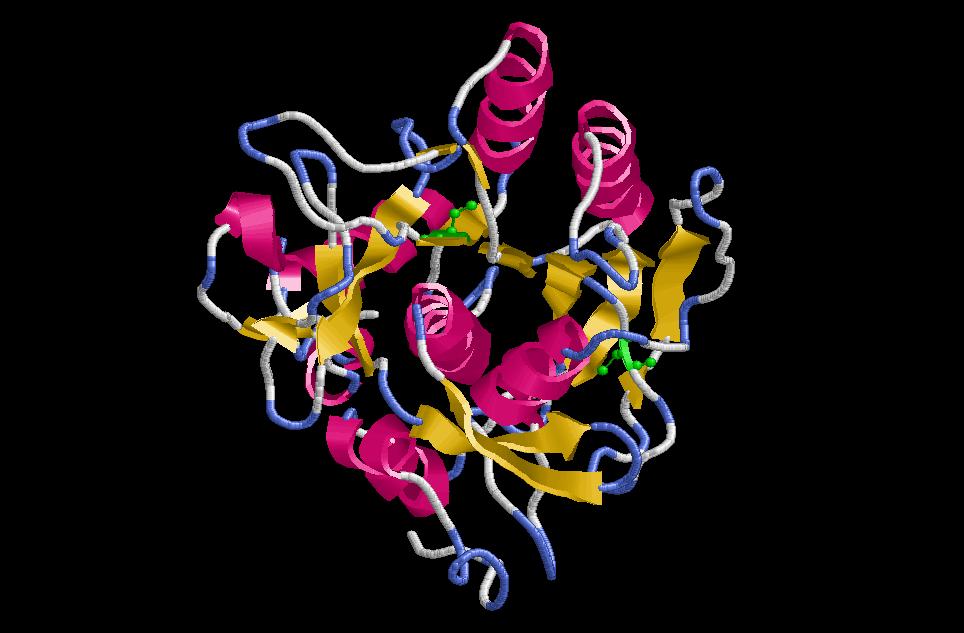
I la distància que hi ha entre aquestes dues cisteïnes marcades en verd ens la calcula el mateix rasmol si li indiquem amb la comanda pick distance i a continuació marquem les dues cisteïnes:
Atom #1: CYS175.CA (1284)
Atom #2: CYS53.CA (403)
Distance CYS175.CA-CYS53.CA: 17.554
Per tant, la dist`ncia entre la cisteïna 175 i la 53 és de 17,554 A (impossible que s'hi formi un pont disulfur!)
C. Per acabar, cridem al programa arrangeG.pl perquè
millori el format del pdb del nostre model, de tal manera que el
en grumos no hi tingui cap problema. Una característica
imprescindible és que l'output ha de ser .pdb
perquè funcioni amb el programa:
$ arrangeG.pl P11018.B99990002_C model_02_C.pdb
2. I comencem la optimització:
] grumos
NAME> /disc9/BE/e16936/practica_7/model_02_C.pdb
(és la ruta d'accés al model)
Give me the directory name: modelet (et demana el nom del
directori on hi guardarà tots el outputs
creats)
Do you need some more files? yes/<no>: no
Do you want to clean the directory you gave me? yes/<no>: no
Options:
- <a> ====> Create INPUTS
- b ====> RUN a process
- c ====> ANALYSIS
- d ====> NEW SIM
- e ====> Logout of this program
Escollim la opció a
Options of Inputs: opció a ====> TOPOLOGY
Options: opció a ====> Single System
TITLE: practiques
A continuació et pregunta si hi ha ponts dissulfur en el
model; però no és el nostre cas. Així doncs a les preguntes referents a això contestarem per defecte, que és no
Options: opció b ====> RUN a process
Options: opció a ====> TOPOLOGY
Options: opció a ====> Single System
Options: opció a ====> Create INPUTS
Options of Inputs: opció b ====> ENERGY OPTIMIZATION
Have you run the TOPOLOGY program? no/<yes>: yes
Options: opció a ====> Single System
Options, value: opció 2 ====> Steepest Descent
Number of steps in each run: 1000
How many times the optimization must be run?: 10
First value of Lambda (parameter) <0.05>: (intro)
Maximum value of Lambda (parameter) <0.10>: (intro)
Options, value: opció 1 ====> SHAKE is not used
Options, value: opció 1 ====> By interaction between groups pair
After how many steps the list change <10>: (intro)
Value of RCUTP <0.8 nm>: (intro)
Value of RSWI2 <10.0 nm>: (intro)
Value of RCUIS <10.0 nm>: (intro)
Value of RCUTL <1.3 nm>: (intro)
Sequence radius to calculate the interaction: 99999
Options, value: opció 1 ====> No periodicity is taked into account
Options, value: opció 1 ====> No position restraining
Options, value: opció 1 ====> No distance restraining
Options, value: opció 1 ====> No dihedral restraining
Print energy every n-steps, n= 100
Options: opció b ====> RUN a process
Options of Inputs: opció b ====> Energy optimization
Options: opció a ====> Single System
Have you run the TOPOLOGY program? no/<yes>: yes
Have you made the DYNAMIC input? no/<yes>: no
Do you want continue with the optimization? no/<yes>: yes
Options: opció a ====> Start the optimization
Options: opció e ====> Logout of this program
3. Ens ha creat una sèrie de directoris (coordinates i
energies) amb molts outputs. Dins de coordinates hi trobarem el
model energèticament optimitzat (model_02_Cxemnum010.gsf)
i a energies hi ha una sèrie de fitxers d'on podrem veure
com ha millorat l'energia potencial a mesura que es feien
més passos de l'optimització. Les energies de l'STEP 100 són les següents:
STEP NIP1 NIS1 NIP2 NIS2 NIP3 NIS3 NITI STEP-SIZE RMS-F-FREE RMS-F-CONS
E-POT-TOT E-BOND-H E-BOND E-ANGLE-H E-ANGLE E-IM-DIH-H E-IM-DIH E-DIH-H E-DIHEDRAL
E-EL-G1-G1 E-EL-G1-G2 E-EL-G2-G2 E-EL-G1-G3 E-EL-G2-G3 E-EL-G3-G3 E-EL-G1-G4 E-EL-G2-G4 E-EL-G3-G4 E-EL-G4-G4
E-LJ-G1-G1 E-LJ-G1-G2 E-LJ-G2-G2 E-LJ-G1-G3 E-LJ-G2-G3 E-LJ-G3-G3 E-LJ-G1-G4 E-LJ-G2-G4 E-LJ-G3-G4 E-LJ-G4-G4
E-DIS-R. E-POS-R. E-DIH-R.
1000 1 0 0 0 0 0 0 0.7856E-03 0.5162E+01 0.5162E+01
-0.17983E+05 0.2018E+02 0.2069E+03 0.2077E+03 0.1512E+04 0.1835E+03 0.3980E+03 0.3950E+02 0.1211E+04
-0.9598E+04 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00
-0.1216E+05 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00
0.0000E+00 0.0000E+00 0.6815-269
És especialment important mirar l'energia potencial total (E-POT-TOT), que normalment comença a l'STEP 1 sent positiva i acaba sent negativa, com era d'esperar. L'altre output que ens interessa és pròpiament el model optimitzat: model_02_Cxemnum010.gsf (dins el directori coordinates).
Com a últims passos de l'anàlisis del model, i
fora del que seria aquesta pràctica pròpiament,
realitzarem un XAM i un procheck amb el model optimitzat:
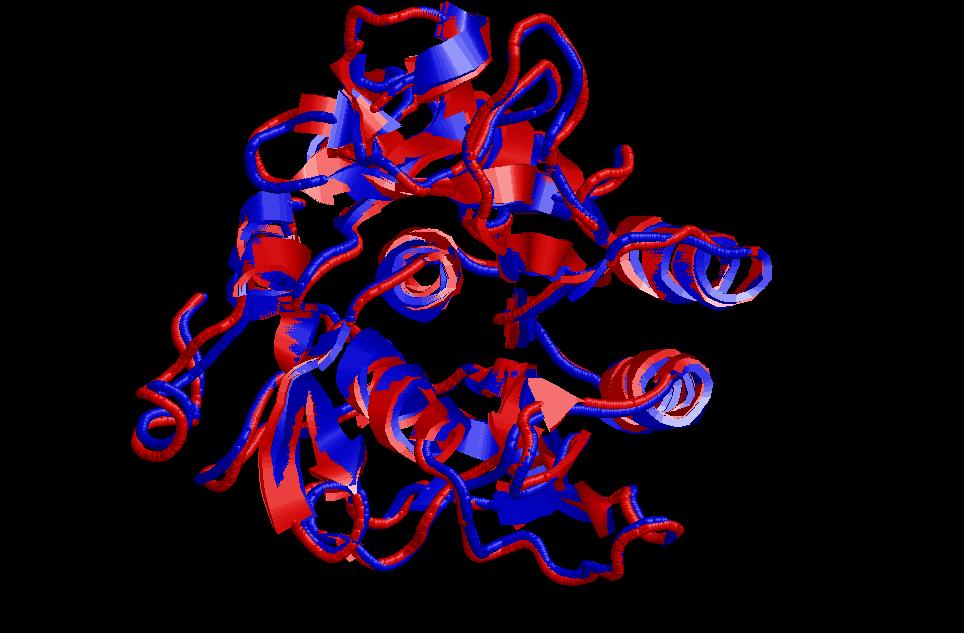
- 1. El XAM es fa entre el model_02_Cxemnum010.gsf i el
model_final.pdb (model abans de ser optimitzat)
- 2. El procheck serà per comprovar que realment ha
millorat, i el compararem amb el procheck del model_02_C.pdb
Del XAM obtenim un pdb de la superposició i també un fitxer output que ens diu el valor de l'RMSD en aquesta superposició:
# RMSD table
#
# 1 2
# 1 0.61
# 2 0.00
I finalment el procheck té el següent resultat:
+----------<<< P R O C H E C K S U M M A R Y >>>----------+
| |
| meu_model.pdb 2.2 304 residues |
| |
*| Ramachandran plot: 70.5% core 25.2% allow 1.9% gener 2.3% disall |
| |
*| All Ramachandrans: 34 labelled residues (out of 302) |
+| Chi1-chi2 plots: 5 labelled residues (out of 169) |
+| Main-chain params: 5 better 0 inside 1 worse |
| Side-chain params: 5 better 0 inside 0 worse |
| |
*| Residue properties: Max.deviation: 5.9 Bad contacts: 0 |
*| Bond len/angle: 5.8 Morris et al class: 2 2 2 |
+| 1 cis-peptides |
+| G-factors Dihedrals: -0.63 Covalent: 0.01 Overall: -0.36 |
| |
| M/c bond lengths: 99.7% within limits 0.3% highlighted |
| M/c bond angles: 92.0% within limits 8.0% highlighted |
*| Planar groups: 76.3% within limits 23.7% highlighted 9 off graph |
| |
+----------------------------------------------------------------------------+
+ May be worth investigating further. * Worth investigating further.
I també podem observar com ha millorat amb el mapa de Ramachandran:
Conclusions finals: Fins aquí hem triat model i l'hem millorat optimitzant-lo. El següent pas seria la simulació per dinàmica molecular però no hem tingut temps de comprovar-ho. Fins aquí ja podem treure conclusions. Aquests últims resultats per evaluar el model ja optimitzat no són molt bons si mirem els resultats del procheck. Encara que el número de bad contacts és 0, el percentatge de residus en zones prohibides del mapa de Ramachandrann és més elevat que quan teníem el model sense optimitzar. El que ens hem de preguntar és si aquesta diferència és important i significativa. La meva conclusiós és que no és despreciable. Potser hi ha més residus en zones no permeses a costa de reduir el número de bad contacts. És a dir, que la optimització ens ha generat un model energ6èticament favorables però estereoquímicament no tant. El xam, de totes maneres, ens demostra la semblança del model optimitzat amb el que partíem abans de fer el grumos. Ens serveix per comprovar que ha canviat, que la optimització ha donat resultat.
Finalment, crec que l'evaluació del meu model hauria d'haver estat més acurada i per millorar ara el model potser hauríem d'anar al templates i buscar-ne algun que acabés de cobrir els gaps interiors de l'alineament.
ÍNDEX