BIPS help
Introduction
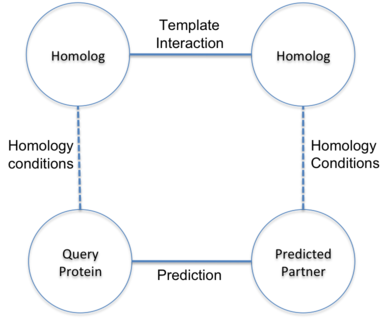
Prediction of interactions inferred by homology. The hypothesis of this approach is that two proteins
(A and B) interact if it exists a known interaction between two proteins (A' and B') such that A is
similar to A' and B similar to B'. The known interactions between A' and B' is called template interaction.
In BIPS server, protein A is the user query protein and protein B is predicted target protein.
The basis of the hypothesis is to assume the similar behavior of homolog proteins.
The approach uses sequence similarity between proteins based on the sequence alignment.
We align the sequences of two proteins to measure their similarity as a function of the
percentage of identical residues and the percentage of their sequence being aligned
(for example, using 60 % identity and 70 % of the total length of the target protein and 90%
of the template). Template sequence coverage is always fixed to 90%.
In a second approach we measure the similarity of the target sequences (A and B) with PFAM domains
as a function of the e-value calculated with the package .
This results in the assignation of one or several PFAM domains to the query and target sequences.
Then, we use the database of iPfam and 3DiD to check for domain-domain interactions.
We hypothesize that A interacts with B if a domain A' can be assigned to A and a domain B' to B
such that A' and B' are interacting domains in iPfam or 3DiD.
Furthermore, it has been shown that the specificity of some interactions depends on a set of
interacting domains. Therefore, the most restrictive set of predictions will be those for which
both criteria of similarity are required, using stringent values of sequence identity, coverage
and domain assignation.
Filters
Predictions can be filtered at different levels:
Filter template interactions
Template interaction detection methods
- Use all methods. Apply "matrix" model to co-complex methods
- Use all methods. Apply "spoke" model to co-complex methods
- Exclude co-complex methods as Tandem Affinity Purification
- Use only interactions reported in at least two different experiments
- Use interactions reported in at least two species
Domain interactions.
We hypothesize that protein A interacts with protein B if a domain A1 can be assigned to A and a domain B1 to B, such that A1 and B1 are interacting domains in iPfam or 3DiD databases. We measure the similarity of the target sequences (A and B) with PFAM domains as a function of the E-value calculated with the package HMMER 3.0. We assign Pfam domains using an E-value cut-off of 10-5 in the Pfam A database.
Filter Homology conditions
Sequence similarity measure. Sequence similarity between proteins relies on BLAST alignments. Query protein sequences are aligned against all sequences with known interactions stored in the BIANA MySQL database. The alignments provide a similarity measure based on the percentage of identical residues aligned (%id) and the percentage of the sequence length of the queries and templates covered by the alignment (query and template coverage, respectively). We use a threshold of 90% of template coverage to ensure that the prediction is not inferred from local regions of the template interaction. BIPS server uses a local database of similarity-measures to avoid unnecessary repeats of BLAST comparisons. This speeds up the server, allowing users to obtain predictions of interactions of full proteomes in manageable time. In this manner, only entirely new sequences consume extra time, and that only in the first run.
Parameters:
- Blast e-value: E-value in the BLAST alignment between target protein and template interacting protein.
- %identities: Identical residues aligned between target protein and template interacting protein.
- Sequence coverage: Percentage of the sequence length of the target protein aligned with template interacting protein.
Clusters of orthologous genes. Two proteins are considered orthologous if they are included in the same cluster of orthologous genes (COG). Orthology definitions between proteins are extracted from eggNOG (30) and Ensembl Compara (31) databases. Our predictions can be filtered assuming the traditional definition of interologs: two target proteins are supposed to interact if they are orthologous with two proteins that interact.
Filter Predicted target proteins by their properties
Partner proteins can be filtered according to:
- Include or exclude specific keywords in their descriptive attributes.
- A specific taxon, including the case in which query proteins are from a particular pathogen and the predicted partners can come from selected hosts
- A subset of proteins uploaded by the user
- Proteins with transmembrane predicted regions (calculated with TMHMM).
Filter predicted interactions by properties of inferred interacting protein pair
- Interacting proteins likely share biological processes or share similar locations compared to non-interacting proteins. Thus, we can use the number of similar functional annotations between the proteins of a predicted interaction to infer the likelihood of the prediction. BIPS uses GO annotations to select the most probable prediction of a query protein by selecting those partners that share the largest number of similar GO terms. The similarity between GO terms implies that either they are equal or there is a parenthood relationship between them.
BIPS predictions for the following species
Here you can find pre-calculated interactomes predicted for some species. You can submit complete proteomes to the BIPS server. We tried to make it as fast as possible. If you submit several thousands of proteins to the server and you do not get your results in less than an hour, please report it at: javier.garciag@upf.edu and we will try to fix it.
Human (TaxID: 9606)
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates.
Download
Arabidopsis (TaxID: 3702)
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates.
Download
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates. Filter to Pfam domain-domain interactions (3DID or iPfam) and GO filter applied at level 3 (BP, CC or MF).
Download
Salmonella (TaxID: 90371)
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates.
Download
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates. Filter to Pfam domain-domain interactions (3DID or iPfam) and GO filter applied at level 3 (BP, CC or MF).
Download
Crosstalk Salmonella-Arabidopsis (90371:3702)
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates.
Download
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates. Filter to Pfam domain-domain interactions (3DID or iPfam) and GO filter applied at level 3 (BP, CC or MF).
Download
Crosstalk Salmonella-Human (90371:9606)
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates.
Download
80% sequence identity, 80% coverage, Joint evalue<1e-80. Exclude Tandem Affinity interactions as templates. Filter to Pfam domain-domain interactions (3DID or iPfam) and GO filter applied at level 3 (BP, CC or MF).
Download
