
 Genes Underlying Inheritance Linked Disorders
Genes Underlying Inheritance Linked Disorders
Introduction
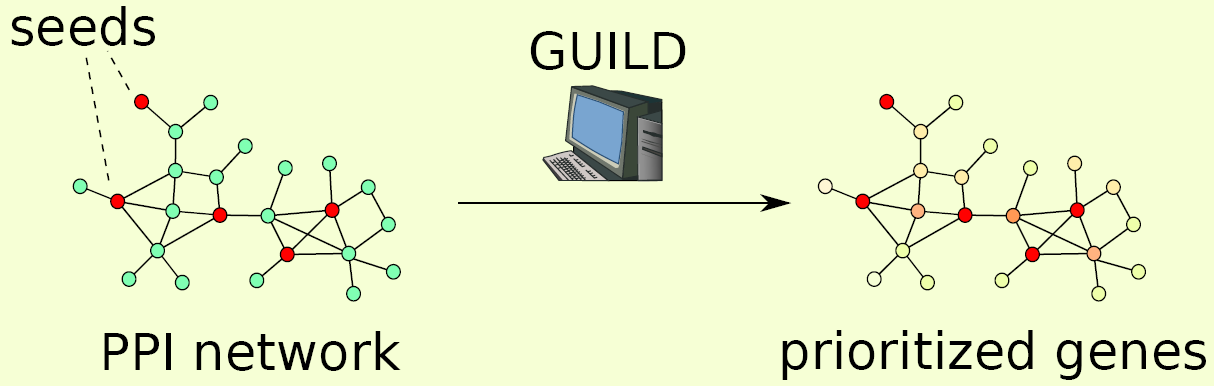
The past decade has witnessed dramatic advances in genome sequencing and a substantial shift in the number of genome wide association studies (GWAS). These efforts have expanded considerably our knowledge on the sequential variations in Human DNA and their consequences on the human biology. Complex genetic disorders often involve products of multiple genes acting cooperatively. Nevertheless, pinpointing the decisive elements of such disease pathways is still a challenge. Recently, network biology has proven its use in identifying candidate genes associated with a disease based on the simple observation that proteins translated by phenotypically related genes tend to interact, so called guilt-by-association principle. Here, we present GUILD (Genes Underlying Inheritance Linked Disorders), a network-based prioritization framework to unveil genes associated with a disease phenotype (disease-genes). In GUILD, we exploit several communication mechanisms between disease-genes emerging from the topology of the interaction network. We used three sources of gene-phenotypic association to specify nodes involved in a disorder (including Online Mendelian Inheritance in Man database)previously published data sets. Analyses on multiple human disease phenotypes demonstrated that the methods proposed in GUILD effectively prioritize genes even when the linkage information is not known a priori. We compared the algorithms we have developed with state-of-the-art prioritization methods such as PageRank with priors, Functional-Flow, Random walk with restart and Network propagation. We also tested the robustness of the approaches proving the effect of the network properties and the independence with the number of original genes/proteins associated with the function or phenotype. Finally, we applied GUILD to prioritize genes in the case of Alzheimer Disease.
Software
GUILD (Genes Underlying Inheritance Linked Disorders) is a framework built for the prioritization of disease candidate genes using a priori gene-disease associations and protein interactions. GUILD consists of implementations of 8 algorithms: NetScore, NetZcore, NetShort, NetCombo, fFlow, NetRank, NetWalk and NetProp. NetScore, NetZcore, NetShort, fFlow and NetRank are implemented in C++ while NetWalk and NetProp are implemented in R and NetCombo is a Python script combining the results of NetScore, NetZcore and NetShort. In this manual, we describe how to use these programs included in GUILD framework. See github repository for the latest version of the code. For Python scripts used to create input files see guild_utilities in toolbox package.
Download
The software used in GUILD framework is distributed freely under GNU Public License. Download the source code, example input files and the software manual here. See handbook either in the downloaded archive (under "doc" directory) or below for the instructions on installation and usage. A PDF version of the handbook is also provided for convenience.Handbook
Contents
1 Requirements
- GCC (GNU project C/C++ compiler) (version 4.3 or higher)
- make (GNU project utility to maintain groups of programs)
- R (version 2.12.1 or higher) (only required for running NetWalk and NetProp)
Unix-like operating systems typically ship with these programs. If not these programs are freely available online. Note that, Windows users can have the fundamental environment for the installation (GCC and make) through MinGW (http:// http://www.mingw.org ) or Cygwin ( http://www.cygwin.com ). R is a free software environment for statistical computing and graphics and available at http://www.r-project.com /.
2 Installation
Download and unpack the source package guild.tar.gz located at http://sbi.imim.es/web/GUILD.php e.g. as follows
{bash}
\$> tar xvzf guild.tar.gz
Then, go to the extracted directory
{bash}
\$> cd guild
Next, go to the src folder and issue make command as below. Beware
that make command in MinGW can have a different name (e.g.
mingw32-make.exe)
MacOS users: use make -f Makefile.mac instead of make command
below (available in the github version of the code).
{bash}
\$> cd src
\$> make
An executable named guild should be created under the ``guild'' folder.
Try running it as follows
{bash}
\$> cd ..
\$> ./guild
If you get the following output when you run it, the installation is successfully
completed.
{bash}
./guild [ Copyleft (GPLv3) - 2011 - Emre Guney (Universitat Pompeu Fabra) ]
Arguments:
-s <prioritization_method>{NetScore:s|NetZcore:z|NetShort:d|fFlow:f|NetRank:r}
-n <node_file>
-e <edge_file>
-o <output_file>
-i <number_of_iterations>
-r <number_of_repetitions>
-t <seed_score_threshold>
-x <number_of_sampled_graphs>
-d <sampled_graph_prefix>
-h
Otherwise make sure that you have recent versions of GCC and
make installed, check the steps above and retry compiling.
If you encounter compilation issues due to BOOST code, try compiling using a newer version of BOOST library (changing the BOOST lib path in the Makefile).
3 Usage
For algorithms implemented in C++, a typical GUILD call consist of several mandatory arguments (such as name of the input/output files and type of the prioritization method) followed by prioritization method specific arguments. Mandatory arguments common to all prioritization methods are explained below, method specific arguments are described in the later sections for each method separately. Possible arguments for a GUILD executable call is as follows:
{bash}
\$> ./guild -s <prioritization_method> -n <node_file> -e <edge_file> -o <output_file>
-i <number_of_iterations> -r <number_of_repetitions> -t <seed_score_threshold>
-x <number_of_sampled_graphs> -d <sampled_graph_prefix>
where;
- prioritization_method:
- The type of the prioritization algorithm,
available values are
- s: NetScore
- z: NetZcore
- d: NetShort
- f: fFlow
- r: NetRank
- node_file:
- Input node scores file containing node (e.g. protein or
gene) identifier followed by its phenotypic relevance score (e.g.
association with the disease phenotype for that protein/gene) on each
line. The values need to be separated by whitespace(s). That is;
{text} <node_id> <node_score> - edge_file:
- Input edge scores file containing node (e.g. protein or
gene) identifier followed by score of the edge its phenotypic
relevance score (e.g. association with the disease phenotype for the
proteins/genes it is connecting) and node identifier (the interaction
partner) on each line. The values are separated by whitespace(s). Thus,
a line in this file looks like;
{text} <node_id> <edge_score> <node_id> - output_file:
- Output node scores file containing node (e.g. protein
or gene) identifier followed by its ``calculated'' phenotypic
relevance score (e.g. association with the disease phenotype for that
protein/gene) on each line. The values are separated by whitespace(s).
The format of a line would be;
{text} <node_id> <node_score>
For algorithms implemented in R (NetWalk and NetProp), a typical GUILD call would look like:
{bash}
\$> R --slave --args <node_file> <edge_file> <output_file> <use_propagation> < random_walk.r
where all arguments are as explained before except ``use_propagation'', which -if provided- converts NetWalk algorithm to NetProp.
3.1 NetScore
NetScore adopts a message-passing scheme such that each node sends the information associated with it as a message to all of its neighbours and the neighbours convey these messages to their neighbours. NetScore takes into consideration alternative shortest paths within the distance of at most number_of_iterations links at each so called repetition-cycle. At the end of the repetition cycle, the node scores are updated according to messages recieved so far and the message passing is restarted.
![\includegraphics[scale=0.5]{netscore.eps}](http://sbi.imim.es/guild/guild-img1.png)
Method specific parameters for NetScore are;
- number_of_repetitions:
- The number of resets (updating the scores of the nodes to calculated scores so far) in the algorithm. Defines the reach of the method (number of links to look further while calculating score) in accordance with the number_of_iteration parameter.
- number_of_iterations:
- Number of iterations to apply the scoring step of the prioritization algorithm. That is the length of shortest paths to consider from a node to other nodes (messages of the nodes that are this many links further are considered).
The following is an example call to run NetScore algorithm using node file node_score.txt and edge file edge_score.txt with number of iteration and repetition parameters of 2 and 3 respectively, writing the calculated scores to a file named output.txt.
{bash}
\$> ./guild -s s -n data/test_proteins.txt -e data/test_interactions.txt -o output.txt -r 3 -i 2
3.2 NetZcore
NetZcore assigns a normalized score using the distribution of the scores of neighbouring nodes. The normalization uses a random model of networks and it is calculated with the Z-score formulae: z=(x-m)/s, where m is the average of scores of neighbouring nodes with similar distribution in the random network and s is the standard deviation. The distribution is obtained with hundred network-replicates obtained by randomly shuffling the scores among nodes with similar degree (i.e. 100 random networks preserving the original topology).
![\includegraphics[scale=0.5]{netzcore.eps}](http://sbi.imim.es/guild/guild-img2.png)
Method specific parameters for NetZcore are;
- number_of_iterations:
- Number of iterations to apply the scoring step of the prioritization algorithm.
- number_of_sampled_graphs:
- Number of the sampled networks (random networks with similar characteristics -e.g. topology or degree distribution- of original network) used for calculating expected mean and standard deviation of scores by random.
- sampled_graph_prefix:
- The full prefix of the sampled networks. An integer from 1 to n_sampled_graphs will be appended at the end of this text while reading sampled networks (thus it should include the directory under which random network files reside). Note that the program itself does not create random networks and looks for already existing random networks residing under the path given by this parameter. A python script is provided for creating such networks (see below).
An example call to run NetZcore algorithm where data folder contains 100 randomly generated networks starting with the prefix test_interactions.txt. (e.g. test_interactions.txt.1, test_interactions.txt.2, ..., test_interactions.txt.100 is as follows:
{bash}
\$> ./guild -s z -n data/test_proteins.txt -e data/test_interactions.txt -o output.txt -i 5
-d data/test_interactions.txt. -x 100
A python script named ``create_random_networks_for_netzcore.py'' is provided
for creating random networks that are going to be used by NetZcore. It requires
Python (version 2.5.2 or higher) and Python NetworkX (version
1.1 or higher) package to be installed in your system. The following command
would create 100 random networks with the same topology of given input network
``data/test_interactions.txt'' with the prefix of
``data/test_interactions.txt.'' (appends a dot at the end of the provided egde
scores file name).
{bash}
\$> python src/create_random_networks_for_netzcore.py data/test_interactions.txt 100
3.3 NetShort
NetShort accumulates the weighted shortest path lengths between a node and the rest of nodes in the network, where each edge-weight is inversely proportional to the average of the scores of the two nodes connected by the edge (i.e.edges connecting high scoring nodes are shorter).
![\includegraphics[scale=0.5]{netshort.eps}](http://sbi.imim.es/guild/guild-img3.png)
There is no method specific parameter for NetShort, however note that
algorithm uses the phenotypic association scores in the edge scores file
(edge_file) rather than the node scores file (e.g. the average of
the scores of the nodes the edge in concern connects). A python script to
create netshort specific edge scores file is provided for convenience (see
below).
Thus an example NetShort run would be;
{bash}
\$> ./guild -s d -n data/test_proteins.txt -e data/test_interactions_for_netshort.txt -o output.txt
A python script named ``convert_network_for_netshort.py'' is provided
for creating edge scores file that is going to be used by NetShort
(where original edge scores are multiplied by average of the scores of the
nodes the edges belong to). It requires Python (version 2.5.2 or
higher). The following command would convert the original edge scores file
``data/test_interactions.txt'' to a NetShort specific
``data/test_interactions_for_netshort.txt'' egde scores file using node
scores information in ``data/test_proteins.txt''.
{bash}
\$> python src/convert_network_for_netshort.py data/test_proteins.txt data/test_interactions.txt
data/test_interactions_for_netshort.txt
3.4 fFlow
In fFlow (based on the algorithm of Functional Flow in Nabieva et al. 2005), at each iteration, annotation scores flow from nodes with higher score towards nodes with lower scores at the amount of the capacity of the edge through which the nodes are connected.
The method specific parameters for fFlow are;
- number_of_iterations:
- Number of iterations to apply the scoring step of the prioritization algorithm.
- seed_score_threshold:
- All the nodes that have higher than this given threshold score will be considered as seeds for the method and assigned infinite scores during scoring.
An example call of fFlow where all nodes that have a score higher than ![]() are seeds is;
are seeds is;
{bash}
\$> ./guild -s f -n data/test_proteins.txt -e data/test_interactions.txt -o output.txt -i 5 -t 1.0
3.5 NetRank
NetRank (based on the ToppGene algorithm proposed by Chen et al. 2009) uses Page Rank with priors algorithm (where a random surfer is more likely to end up in initially relevant nodes) to score a node in terms of phenotypic relevance. The damping factor is 0.15 and the number iterations for convergence is defined by the number_of_iterations parameter (see below).
The method specific parameter for NetRank is;
- number_of_iterations:
- Number of iterations to apply the scoring step of the prioritization algorithm. In the case of Page Rank algorithm it defines the number of the times for calculating page ranks for the nodes (convergence criterion for the algorithm). This number is set to 20 by default.
Therefore an example run for NetRank is as follows:
{bash}
\$> ./guild -s r -n data/test_proteins.txt -e data/test_interactions.txt -o output.txt
3.6 NetWalk
NetWalk (based on the Random walk with restarts algorithm proposed by Kohler, et al., 2008) iteratively simulates random transitions of a walker from a node to a randomly selected neighbour node and where at any time step the walk can be restarted depending on a predefined probability. Random walk with restarts is slightly different than PageRank with priors in the way that it normalizes the link weights. The convergence is decided by either having a probability difference less than 10e-6 between two consecutive time steps or achieving the limit of the number of iterations, set to 50 (though in practice less than 20 iterations are typically sufficient to satisfy the first criterion). There is no method specific parameter for NetWalk.
Therefore an example run for NetWalk is as follows:
{bash}
\$> R --slave --args data/test_proteins.txt data/test_interactions.txt output.txt < random_walk.r
3.7 NetProp
NetProp (based on the Network propagation algorithm proposed by Vanunu, et al., 2010) modifies random walk with restarts such that the link weight is normalized not only by number of outgoing edges but also by number of incoming edges. The convergence is decided as it is done for NetWalk. There is no method specific parameter for NetProp.
Therefore an example run for NetProp is as follows:
{bash}
\$> R --slave --args data/test_proteins.txt data/test_interactions.txt output.txt 1 < random_walk.r
3.8 NetCombo
NetCombo combines the output scores from NetScore, NetZcore and NetShort in a consensus scheme by averaging normalized scores (z-scores) of a node in all of these methods. It requires the output files of NetScore, NetZcore and NetShort.
Therefore an example run for NetCombo is as follows:
{bash}
\$> python src/combine_scores.py output_netscore.txt output_netzcore.txt output_netshort.txt output.txt