En este caso apareció la proteína de unión a la vitamina D (1J78, 1J7E). La secuencia original que se intentaba modelar era la del precursor de la DBP, por lo que se pensó que estas secuencias serían buenos templates.El alineamiento de esta secuencia con las obtenidas por la búsqueda fue realizado mediante el programa CLUSTALW. Se observó que la única zona no homóloga eran los primeros 17 aminoácidos, que no aparecen en la forma madura de DBP al tratarse de una secuencia señal. Por este motivo, esta región fue cortada, por lo que la secuencia problema y los templates quedaron con una homología del 100%, a excepción de un gap de 7 aminoácidos (96-102) que se corresponde con una región no resuelta de la estructura cristalográfica.
La DBP está formada por 2 cadenas, DBPA y DBPB, que no son totalmente idénticas entre si (rmsd entre A y B:1.73). Se descartó el uso de la cadena A porque en el alineamiento secuencial se observaba un gap más.
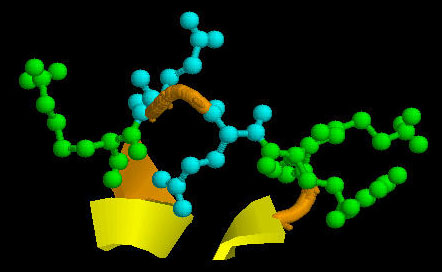
En el caso de las cadenas A, se observó que una de ellas había sido cristalizada acomplejada con vitamina D y la otra no. Se realizó, entonces, un alineamiento mediante el programa XAM y posterior visualización con RASMOL, para comprobar si la unión de la vitamina D provocaba un cambio de conformación en la molécula de DBP, en especial en el dominio I por ser la zona de unión a la vitamina D, pero no se observa ningún cambio apreciable de conformación (Fig 6); se demostró numéricamente calculando el la desviación rms de las dos cadenas obteniéndose un valor de 0.52 Å,muy parecido al existente entre las cadenas B (rmsd:0.59 Å), ambas unidas a vitamina D.
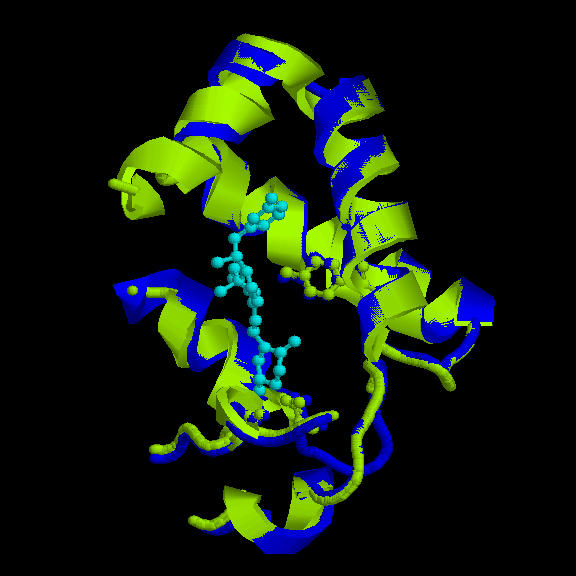
Fig 6. Superposición subdominio Ia de cadenas A
El paso siguiente fue modelar la proteína problema, utilizándose como templates las cadenas B de 1J78 y 1J7E de estructura cristalográfica conocida, aún cuando la secuencia problema y la secuencia de los templates son idénticas. El modelado se realizó mediante el programa MODELLER, basado en restricciones de distancias extraídas del alineamiento múltiple de las secuencias. El modelo resultante fue analizado energéticamente con los programas PROSAII y PROCHECK. Los resultados, como era de esperar, eran muy favorables. Así, un 94.7% de los aminoácidos están situados en el core y no hay ningún mal contacto entre cadenas laterales.
En la estructura cristalográfica de los templates, hay una zona, desde el aminoácido 96 al 102, que no se ha resuelto su estructura, por lo que en el modelo aparece como un loop. Se intentó modelar esta zona con el programa ARCH TYPE que utiliza una base de datos de loops de estructura determinada. Pero no fue posible, ya que no coincidía con ninguno de los determinados en la base de datos.
Se decidió optimizar el modelo mediante el programa GROMOS . El modelo optimizado obtenido se analizó también energéticamente mediante PROSAII y PROCHECK (resultados) Aunque el porcentaje de los aminoáciods en el core es algo menor (89.1%), la energía global del sistema ha disminuído ligeramente, partiendose de un valor inicial de -0.27E+5, se alcanzó el valor de -0.29E+5.El análisis comparativo del perfil de pseudo-energías del template, modelo sin optimizar y optimizado muestra que es idéntico (Fig 7). No se observan zonas de mala energía, aunque la zona N-terminal, que se corresponde con el dominio de unión a la vitamina D, tiene energía algo superior al resto.
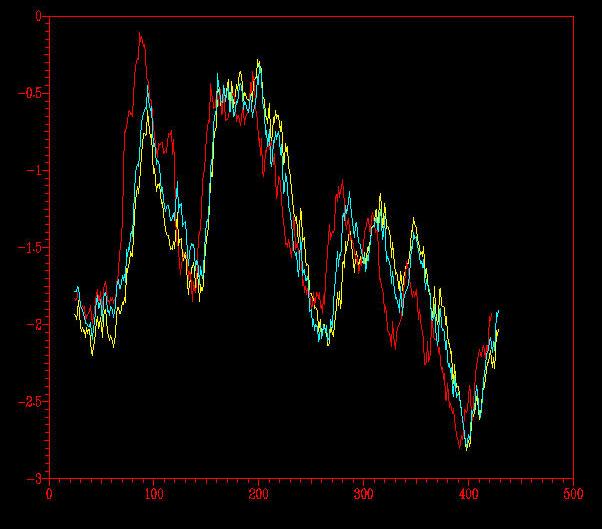
Fig 7. Resultados PROSA. En rojo,template (1J78) en amarillo, modelo optimizado y en cian, modelo no optimizado.
El cálculo de la desviación rms entre el template y el modelo obtenido es de 0.69 Å, lo que evidencia que no existe diferencia estructural entre ambas. No hay que obviar que ambas son la misma proteína.
Dinámica Molecular:
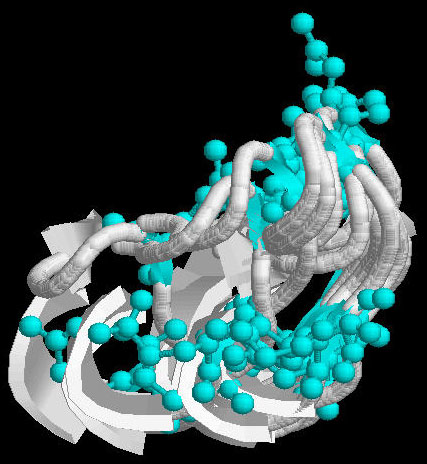
En la estructura de los templates había un gap (aminoácidos 96-102), que en el modelo aparece como un loop. Se realizó la dinámica molecular del sistema mediante el programa GROMOS, para ver la movilidad de la molécula y, en especial, de la región correspondiente al gap.
El loop está entre las hélices 5 y 6 del dominio I, concretamente entre las cisteínas 93 y 104, que forman un puente disulfuro entre sí; este hecho provoca que los extremos del loop tengan muy poca movilidad, pudiéndose mover libremente el centro. Analizando la secuencia de esta zona se observa que está formada fundamentalmente por aminoácidos cargados; concretamente hay una lisina (carga +) y un ácido glutámico (carga -) seguidas y separadas por una glicina y una leucina de otro ácido glutámico, una lisina y un arginina (Fig 8).
Por tanto, se forman claramente dos dipolos con sus cargas negativas orientadas hacia el centro del loop, por lo que estarán en continuo movimiento; este hecho se agudiza por la presencia del solvente.El valor del B-factor no es muy elevado; este hecho puede ser debido a que la dinámica se ha realizado en un medio sin agua, que es donde los dipolos tendrían más fuerza y, por tanto, más movimiento (Fig 9).Fig 8. Loop correspondiente a la región 96-102 (en verde aa con carga positiva, en azul los negativos)
Fig 9. Dinámica molecular de la región 96-102 (en azul aa con carga negativa)
Home | Abstract | Características estructurales | Metodología 1 | Conclusiones | Bibliografía