Objectius: Aquesta pràctica consta de dos parts. Part1: Bàsicament utilitzem els resultats de la pràctica anterior (alineament amb ClustalW) per fer un alineament estructural amb el programa STAMP (està explicat més detalladament a la pràctica 4.2). Part2: Generem un perfil de Hidden Markov amb els mateixos templates i tornem a alinear les seqüències. A partir d'aquí, tots els passos que fem en les altres pràctiques seran dobles: un amb l'alineament generat per clustalw i l'altre pel de HMM
Primera PART:
1. Treballarem en tcsh en comptes de bash perquè la
crida de programes és més ràpida i
còmode:
$ tcsh
] source /disc9/cshrc
2. Hem d'estar localitzats al mateix directori que contingui
els pdb dels templates escollits:
] kwrite model.domains
És necessari crear un fitxer .domains on hi
especifiquem el nom de la proteïna que estarà escrit
en l'alineament i la cadena amb les que es farà l'stamp
(si la proteïna en té més d'una). Primer, se li diu la ruta on tribarà el pdb, després el nom que li direm a aquest i tot seguit si és tota la proteïna ({ALL}) o només una cadena ({chain A}):
./pdb1scj.pdb 1scjA {chain A}
./pdb1tec.pdb 1tecE {chain E}
./pdb1yjc.pdb 1yjc {ALL}
./pdb1ak9.pdb 1ak9 {ALL}
3. Cridem l'stamp:
] stamp -l model.domains -rough -n 2 -prefix model
4. El resultat serà el model.3, ja que tenim 4 pdb i
genera tants outputs com proteïnes hi ha menys 1.
Això s'explica perquè el programa STAMP fa
alineaments de dos en dos, és a dir, comença
alineant les dues seqüències més semblants
(aquest resultat està en model.1). Seguidament
alinea aquest resultat amb una de les dues seqüències
restants, la que hi tingui més homologia (això ho
emmagatzema a model.2). I així successivament. En
el nostre cas només disposem d'una seqüència
més, és a dir que l'output que conté les
quatre seqüències alineades és
model.3. Es tracta d'un alineament vertical que haurem de convertir.
] kwrite model.3
... RVVV ???? 0 0.00087 2.65242 0.22400 QPPP ???? 0 0.25081 1.37378 2.96500 YYYY ???? 1 0.87582 0.69602 9.81900 GGGG ???? 1 0.94685 0.64419 10.59800 PVIV ???? 1 0.92387 0.78782 10.34600 QSSS ???? 1 0.91685 0.91607 10.26900 KQQQ ???? 1 0.93883 0.84781 10.51000 IIII ???? 1 0.94384 0.78218 10.56500 QKKK ???? 1 0.93983 0.86026 10.52100 AAAA ???? 1 0.94083 0.83947 10.53200 PPPP ???? 1 0.93682 0.83818 10.48800 QAAA ???? 1 0.88183 0.95640 9.88500 ALLL ???? 1 0.74487 1.39487 8.38300 WHHH ???? 1 0.73384 1.29815 8.26200 DSSS ???? 1 0.55784 1.71699 6.33200 ...
5. Ho convertim per visualitzar-ho millor:
] aconvertMod2.pl -in b -out c < model.3 >> resultat
A resultat hi tenim un alineament com aquest:
CLUSTAL W(1.60) multiple sequence alignment 1tecE YTPNDPYFSSRQYGPQKIQAPQAWD-IAEGSGAKIAIVDTGVQSNHPDLAGKVVGGWDF- 1ak9 ----AQS---VPYGVSQIKAPALHSQGYCGSNVKVAVIDSGIDSSHPD-L-KVAGGASFV 1scjA ----AQS---VPYGISQIKAPALHSQGYTGSNVKVAVIDSGIDSSHPD-L-NVRGGASFV 1yjc ----AQS---VPYGVSQIKAPALHSQGYTGSNVKVAVIDSGIDSSHPD-L-KVAGGASFV 1tecE -VDNDST-PQNGNGHGTHCAGIAAAVTNNSTGIAGTAPKASILAVRVLDNSGSGTWTAVA 1ak9 PSE--TNPFQDNNSHGTHVAGTVAAL-NNSIGVLGVAPCASLYAVKVLGADGSGQYSWII 1scjA PSE--TNPYQDGSSHGTHVAGTIAAL-NNSIGVLGVSPSASLYAVKVLDSTGSGQYSWII 1yjc PSE--TNPFQDNNSHGTHVAGTVAAL-DNSIGVLGVAPSASLYAVKVLGADGSGQYSWII 1tecE NGITYAADQGAKVISLSLGGTVGNSGLQQAVNYAWNKGSVVVAAAGNAG----NTAPNYP 1ak9 NGIEWAIANNMDVINMSLGGPSGSAALKAAVDKAVASGVVVVAAAGNEGTSGSSSTVGYP 1scjA NGIEWAISNNMDVINMSLGGPTGSTALKTVVDKAVSSGIVVAAAAGNEGSSGSTSTVGYP 1yjc NGIEWAIANNMDVINMSLGGPSGSAALKAAVDKAVASGVVVVAAAGNEGTSGSSSTVGYP 1tecE AYYSNAIAVASTDQNDNKSSFSTYGSVVDVAAPGSWIYSTYPTSTYASLSGTSMATPHVA 1ak9 AKYPSVIAVGAVDSSNQRASFSSVGPELDVMAPGVSIQSTLPGNKYGAKSGTSMASPHVA 1scjA AKYPSTIAVGAVNSSNQRASFSSAGSELDVMAPGVSIQSTLPGGTYGAYNGTCMATPHVA 1yjc AKYPSVIAVGAVDSSNQRASFSSVGPELDVMAPGVSICSTLPGNKYGAYSGTSMASPHVA 1tecE GVAGLLAS-Q-GRSASNIRAAIENTADK-ISGTGTYWAKGRVNAYKAVQY 1ak9 GAAALILSKHPNWTNTQVRSSLENTTTKLGLGDSFYYGKGLINVQAAAQ- 1scjA GAAALILSKHPTWTNAQVRDRLESTATY--LGNSFYYGKGLINVQAAAQ- 1yjc GAAALILSKHPNWTNTQVRSSLENTTTY--LGDSFYYGKGLINVQAAAQ-
] transform -f model.4 -g -o model.3.pdb
I amb aquesta última comanda hem generat el pdb d'aquest
alineament que el visualitzarem amb el programa rasmol:
] rasmol model.3.pdb
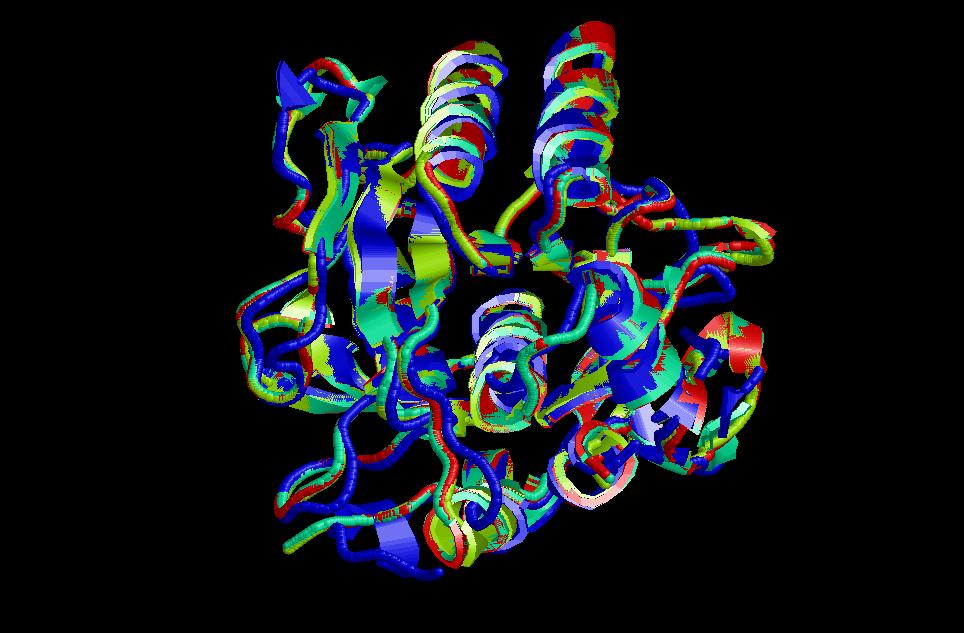
Conclusions: Veiem la superposició de les quatre estructures. És un bon resultat; tant sols en algun loop es veu pobre la superposició. Les hèlix alfa són les zones on més bé, s'ha fet la superposició i això és perquè són les zones més conservades. A més a mé les 4 seqüències són bastant homòlogues entre elles, com es veu en l'alineament. Per tot això, era d'esperar el resultat.
Segona PART:
1. Modifiquem lleugerament l'output resultats de la
primera part. Se li elimina la part dels interrogants i es deixa
només l'alineament en si. Tot seguit, es genera un model
de hidden Markov:
] hmmbuild perfil5.hmm resultat ] hmmalign -o resultat_final.aln
perfil5b.hmm llistat.fa
2. El resultat té l'extensió .aln i per
tant tindrà el mateix format que l'alineament llistat.aln
(resultat del clutalw de la pràctica 5.1)
] emacs resutat_final.aln
# STOCKHOLM 1.0 #=GF AU HMMER 2.2g P11018 mngeirlipYVTNEQIMDVneLPEGIKVIKAPEMWAKGVKGKNIKVAVLD 1ak9 .........----AQS---..VPYGVSQIKAPALHSQGYCGSNVKVAVID 1scjA .........----AQS---..VPYGISQIKAPALHSQGYTGSNVKVAVID 1tecE .........YTPNDPYFSS..RQYGPQKIQAPQAWD-IAEGSGAKIAIVD 1yjc .........----AQS---..VPYGVSQIKAPALHSQGYTGSNVKVAVID #=GC RF .........xxxxxxxxxx..xxxxxxxxxxxxxxxxxxxxxxxxxxxxx P11018 TGCDTSHPDLKNQIIGGKNF--TDDDggkEDAISDYNGHGTHVAGTIAAN 1ak9 SGIDSSHPD-L-KVAGGASFVPSE--...TNPFQDNNSHGTHVAGTVAAL 1scjA SGIDSSHPD-L-NVRGGASFVPSE--...TNPYQDGSSHGTHVAGTIAAL 1tecE TGVQSNHPDLAGKVVGGWDF--VDND...ST-PQNGNGHGTHCAGIAAAV 1yjc SGIDSSHPD-L-KVAGGASFVPSE--...TNPFQDNNSHGTHVAGTVAAL #=GC RF xxxxxxxxxxxxxxxxxxxxxxxxxx...xxxxxxxxxxxxxxxxxxxxx P11018 -DSNGGIAGVAPEASLLIVKVLgGENGSGQYEWIINGINYAVEQKVDIIS 1ak9 -NNSIGVLGVAPCASLYAVKVL.GADGSGQYSWIINGIEWAIANNMDVIN 1scjA -NNSIGVLGVSPSASLYAVKVL.DSTGSGQYSWIINGIEWAISNNMDVIN 1tecE TNNSTGIAGTAPKASILAVRVL.DNSGSGTWTAVANGITYAADQGAKVIS 1yjc -DNSIGVLGVAPSASLYAVKVL.GADGSGQYSWIINGIEWAIANNMDVIN #=GC RF xxxxxxxxxxxxxxxxxxxxxx.xxxxxxxxxxxxxxxxxxxxxxxxxxx P11018 MSLGGPSDVPELKEAVKNAVKNGVLVVCAAGNEG-DGDERTeeLSYPAAY 1ak9 MSLGGPSGSAALKAAVDKAVASGVVVVAAAGNEGTSGSSST..VGYPAKY 1scjA MSLGGPTGSTALKTVVDKAVSSGIVVAAAAGNEGSSGSTST..VGYPAKY 1tecE LSLGGTVGNSGLQQAVNYAWNKGSVVVAAAGNAG----NTA..PNYPAYY 1yjc MSLGGPSGSAALKAAVDKAVASGVVVVAAAGNEGTSGSSST..VGYPAKY #=GC RF xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx..xxxxxxx P11018 NEVIAVGSVSVARELSEFSNANKEIDLVAPGENILSTLPNKKYGKLTGTS 1ak9 PSVIAVGAVDSSNQRASFSSVGPELDVMAPGVSIQSTLPGNKYGAKSGTS 1scjA PSTIAVGAVNSSNQRASFSSAGSELDVMAPGVSIQSTLPGGTYGAYNGTC 1tecE SNAIAVASTDQNDNKSSFSTYGSVVDVAAPGSWIYSTYPTSTYASLSGTS 1yjc PSVIAVGAVDSSNQRASFSSVGPELDVMAPGVSICSTLPGNKYGAYSGTS #=GC RF xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx P11018 MAAPHVSGALALIksyeeesfqrkLSES-EVFAQLIRRTLPLDIAKTLAG 1ak9 MASPHVAGAAALI...........LSKHPNWTNTQVRSSLENTTTKLGLG 1scjA MATPHVAGAAALI...........LSKHPTWTNAQVRDRLESTATY--LG 1tecE MATPHVAGVAGLL...........AS-Q-GRSASNIRAAIENTADK-ISG 1yjc MASPHVAGAAALI...........LSKHPNWTNTQVRSSLENTTTY--LG #=GC RF xxxxxxxxxxxxx...........xxxxxxxxxxxxxxxxxxxxxxxxxx P11018 NGFlYLTAPDELAEKAEQshlltl 1ak9 DSF.YYGKGLINVQAAAQ...... 1scjA NSF.YYGKGLINVQAAAQ...... 1tecE TGT.YWAKGRVNAYKAVQy..... 1yjc DSF.YYGKGLINVQAAAQ...... #=GC RF xxx.xxxxxxxxxxxxxx...... //
Conclusions:Aquest alineament és una mica diferent del obtingut en la primera part però tampoc tant. Aquestes petites diferències, però, faran que generem en les pròximes pràctiques quatre models per a una mateixa proteïna. I això ens permetrà escollir el millor i evaluar els mètodes més convenients en cada cas. A partir d'aquí treballarem amb aquests dos fitxers: llistat.aln (ClustalW) i resultat_final.aln (HMM)