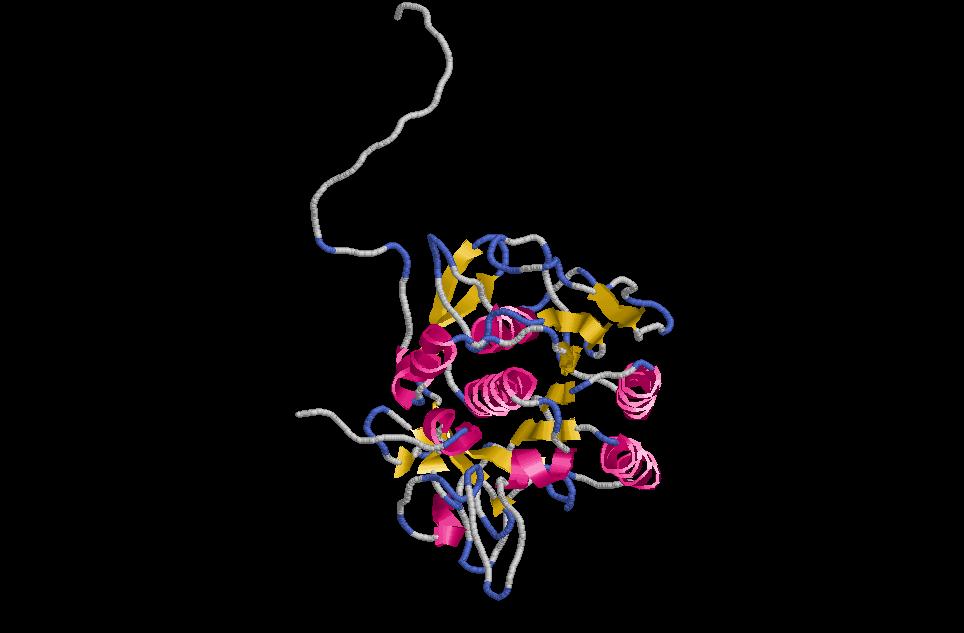
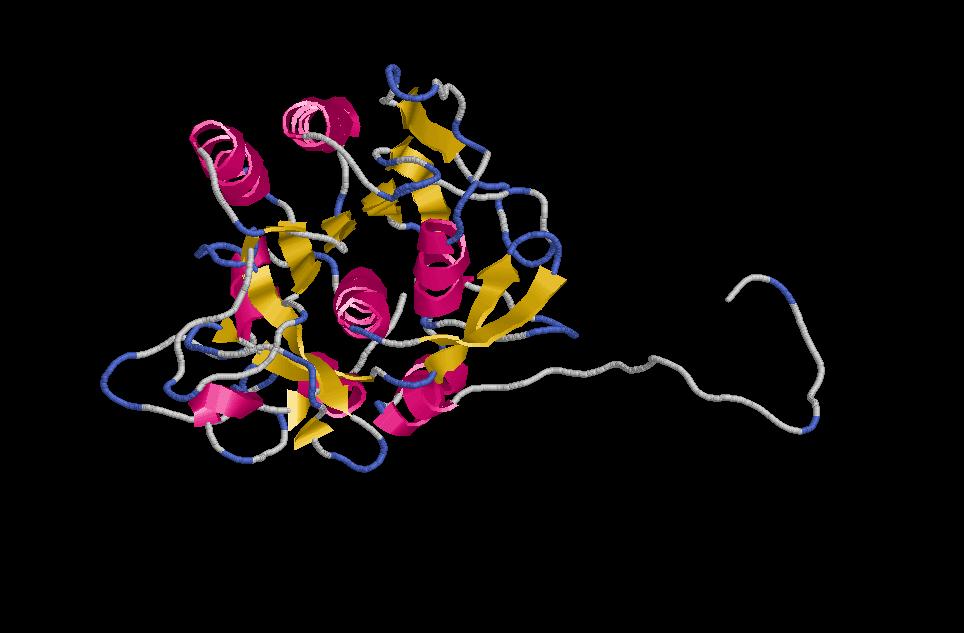
Objectius: Aquesta pràctica es basarà en el programa MODELLER per generar un model estructural únic a partir del alineaments de les pràctiques 5.1 (de clustalw) i 5.2 (de Hidden Markov)
MODELLER: És un programa utilitzat pel modelatge comparatiu de l'estructura tridimensional de proteïnes. L'usuari hi entra un alineament d'una seqüència per ser modelada amb d'altres amb estructura coneguda. Automàticament, el programa calcula un model que conté tots els àtoms excepte els H. Per fer el modelatge té en compte sobretot les restriccins especials dels diferents conjunts d'àtoms, a part de poder fer tasques addicionals que inclouen el modelatge de novo de loops, optimització de varis models de estructura de proteïnes respecte una funció concreta, alineament múltiple de seqüències protèiques i/o d'estructura, clustering, búsqueda de seqüències en databases, comparació d'estructures de proteïnes, etc. La idea bàsica de funcionament d'aquest programa l'explicaré, per simplificar, com si només partíssim d'un template i una seqüència problema:
Exemple:
1. Per conèixer el programa i poder-lo aplicar amb els
nostres models, primer el cridem amb un exemple:
$ tcsh
] source /disc9/cshrc
] cp /disc9/practica_5/subtilisin.tar .
] tar xvf subtilisin.tar (descomprimim l'arxiu)
] cd subtilisin
] ls
2. En el directori subtilisin hi trobem una sèrie de
pdb (1sbh.pdb i 1acjA.pdb), un alinemanet (P7.aln), la
seqüència problema, en format FASTA, amb la que hem
estat treballant fins ara (P11018.seq), i un fitxer
d'extensió .top (P7.top). El que més ens
interessa és aquest últim. L'obrim per veure'n el
seu contingut:
] kwrite p7.top
# PRIMER: STEP 5 # # This script should produce two models, 1fdx.B999901 and 1fdx.B999902. # # Before you run this script, do this: ln alignment.seg.ali fer2.ali # INCLUDE # Include the predefined TOP routines SET ALNFILE = 'p7.ali' # alignment filename SET KNOWNS = '1scjA' '1sbh' # codes of the templates SET SEQUENCE = 'P11018' # code of the target SET ATOM_FILES_DIRECTORY = './' # directories for input atom files SET STARTING_MODEL= 1 # index of the first model SET ENDING_MODEL = 2 # index of the last model # (determines how many models to calculate) SET DEVIATION = 2.0 # have to be >0 if more than 1 model SET RAND_SEED = -12312 # to have different models from another TOP file CALL ROUTINE = 'model' # do homology modelling
En l'apartat Set alnfile hi especificarem el fitxer de l'alineament; és important que estigui en extensió .ali. L'apartat set knows és on s'hi ha d'especificar el
nom de les estructures templates. El següent apartat, set
sequence es refereix a la seqüència problema (que,
tant per l'exemple de la pràctica com per els nostres
models, serà P11018.seq). Els apartats Set Starting_Model i Set ending_model fa referència al número del fitxer que conté el primer i l'últim model. El set deviation indica la diferència de les
distàncies d'un carboni alfa respecte un residu de cada
template (d1-d2>0; sent d1 i
d2 les distàncies entre un C alfa concret i el
residu en posició x de la seqüència template 1
i 2, respectivament).Ha de ser major que 0 si estem treballant amb més d'un model, com és el cas.
Aquest fitxer també ens adverteix que els identificadors
de les seqüències en l'alineament (fitxer P7.aln) no
poden ser del format: SP|P11918|, per exemple, perquè el
programa no ho sap llegir i dóna error.
3. Abans de córrer el modeller fem un SplitChain dels
pdb templates per separar-los en les diferents cadenes i convertim el fitxer de l'alineament a una
extensió .ali:
] PDBtoSplitChain.pl -i pdb1sbh.ent -o 1sbh
] PDBtoSplitChain.pl -i pdb1scj.ent -o 1scj
] aconvertMod2.pl -in c -out p < P7.aln > P7.ali
Si obrim P7.ali veurem que, de fet, és molt diferent a l'alineament que teníem abans. Ara és molt més semblant a un format fasta, però també t'ensenya els gaps de l'alineament. La primera línia amb la que comença cada seqüència ens dóna certa informació: StructureX:1scjA:1 275... i això significa que l'alineament està fet des de l'àtom 1 al 275.
4. Ara ja podem córrer el programa:
] mod p7.top
5. Tenim molts fitxers de sortida:
P11018.B99990001Els que ens interessen per continuar fent el model estructural de la nostra proteïna problema són dos: P11018.B99990001 i P11018.B99990002. Aquests dos es poden obrir amb el rasmol i observem el següent:
P11018.B99990002
P11018.V99990001
P11018.V99990002
P11018.D00000001
P11018.D00000002
...
Interpretació: amb aquesta imatge podem veure si els gaps han donat lloc a problemes de predicció d'estructura. Els gaps intermedis que no siguin coberts per cap seqüència template seran loops que poden trencar l'estructura secundaria (per exemple, poden estar al mig d'una làmina beta o una hèlix alfa). La optimització màxima seria buscar nous templates que cobrissin aquells gaps. Si no és possible de cap manera s'intenta fer córrer els gaps cap als extrems de la proteïna o bé en els loops propis de l'estructura. En les zones que no estan cobertes per templates, el programa no reconeix cap estructura secundària i per tant i introdueix un loop. Com era d'esperar això es veu clarament als extrems. Aquesta cua sense estructura secundària ja la preveiem mirant l'alineament.
Model:
A. ClustalW:
1. L'únic que hem de fer per aplicar-ho al model
és retocar el fitxer p7.top de la següent manera:
# PRIMER: STEP 5
#
# This script should produce two models, 1fdx.B999901 and 1fdx.B999902.
#
# Before you run this script, do this: ln alignment.seg.ali fer2.ali
#
INCLUDE # Include the predefined TOP routines
SET ALNFILE = 'llistat.ali' # alignment filename
SET KNOWNS = '1scjA' '1ak9' '1tecE' '1yjc' # codes of the templates
SET SEQUENCE = 'P11018' # code of the target
SET ATOM_FILES_DIRECTORY = './' # directories for input atom files
SET STARTING_MODEL= 1 # index of the first model
SET ENDING_MODEL = 2 # index of the last model
# (determines how many models to calculate)
SET DEVIATION = 2.0 # have to be >0 if more than 1 model
SET RAND_SEED = -12312 # to have different models from another TOP file
CALL ROUTINE = 'model' # do homology modelling
2. Visualitzem els resultats finals:
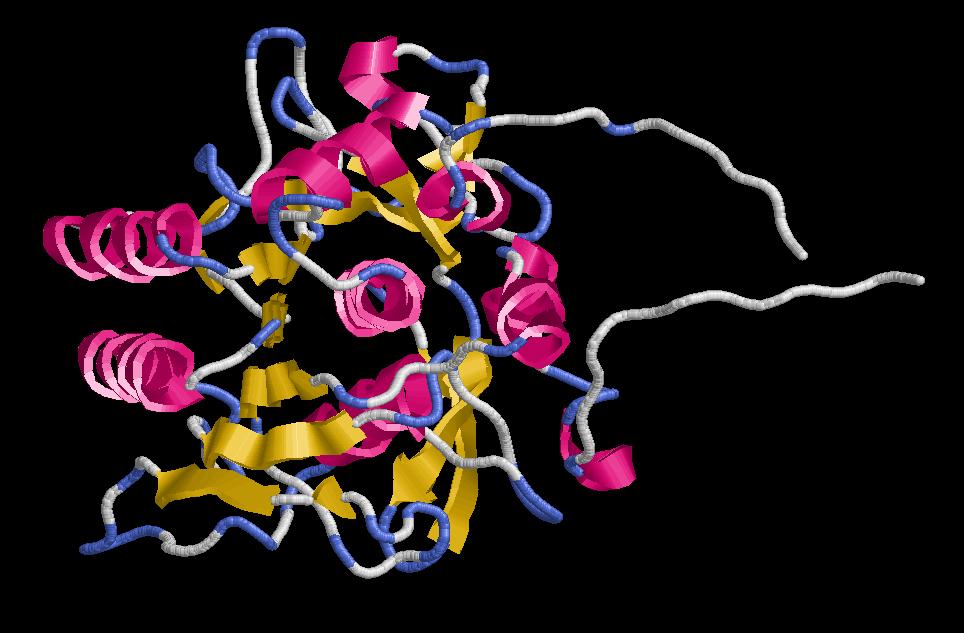
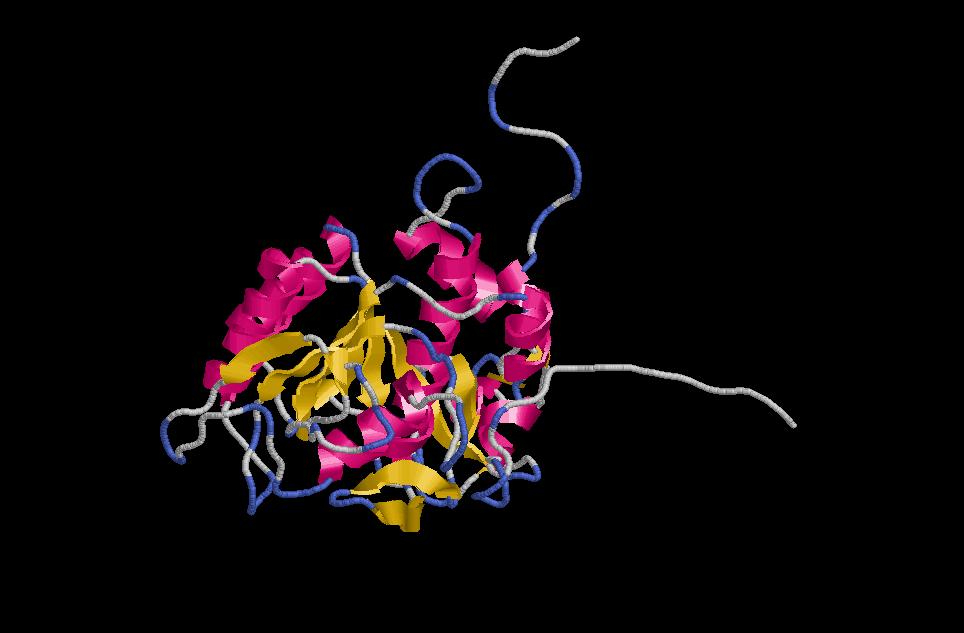
Són els P11018.B99990001 i P11018.B99990002, respectivament. Es poden veure lleugeres diferències entre un i altre. A més a més, aquests dos loops finals sense estrustura secundària ens indiquen que haurem de fer alguns retorcs a l'alineament per millorar el model, perquè simplement no serà gens favorable energèticament. Això ja ho comprovarem més endavant amb el programa PROSA.També hi ha algun loop que trenca alguna estructura secundària i que deixa fragments d'hèlix alfa o làmina beta molt petits. Això és degut als gaps de l'alineament.
B.Hidden Markov:
1. Les mateixes modificacions que hem fet abans són
necessàries aquí i l'única diferència
està en el nom del fitxer que conté l'alineament
(resultat_final.ali), que també hem hagut de convertir de
l'extensió .aln inicial, com a resultat de la pràctica 5.2.
# PRIMER: STEP 5 # # This script should produce two models, 1fdx.B999901 and 1fdx.B999902. # # Before you run this script, do this: ln alignment.seg.ali fer2.ali # INCLUDE # Include the predefined TOP routines SET ALNFILE = 'resultat_modificat_final.ali' # alignment filename SET KNOWNS = '1scjA' '1ak9' '1tecE' '1yjc' # codes of the templates SET SEQUENCE = 'P11018' # code of the target SET ATOM_FILES_DIRECTORY = './' # directories for input atom files SET STARTING_MODEL= 1 # index of the first model SET ENDING_MODEL = 2 # index of the last model # (determines how many models to calculate) SET DEVIATION = 2.0 # have to be >0 if more than 1 model SET RAND_SEED = -12312 # to have different models from another TOP file CALL ROUTINE = 'model' # do homology modelling
2. Visualitzem el resultat:
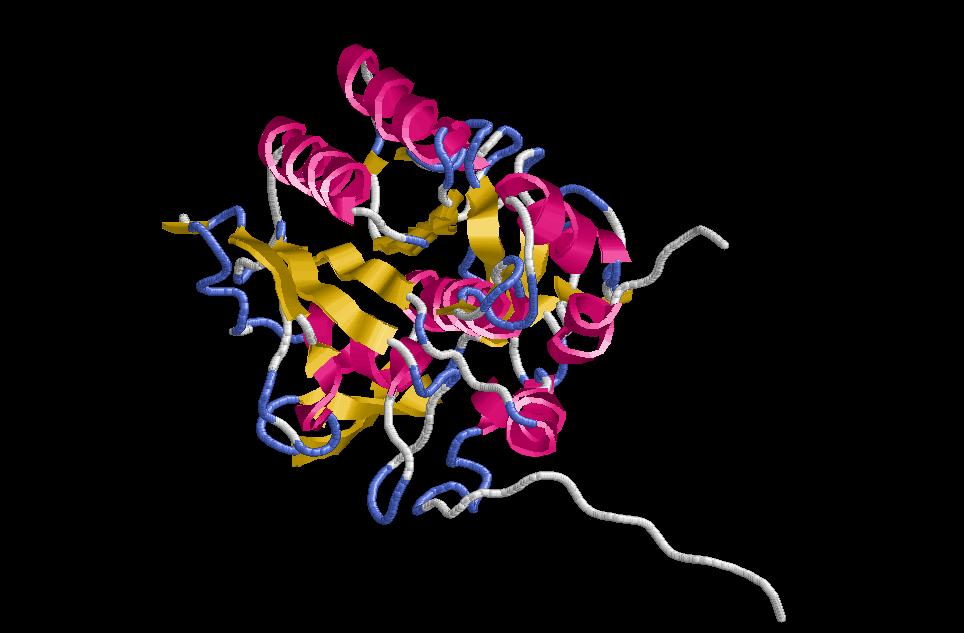
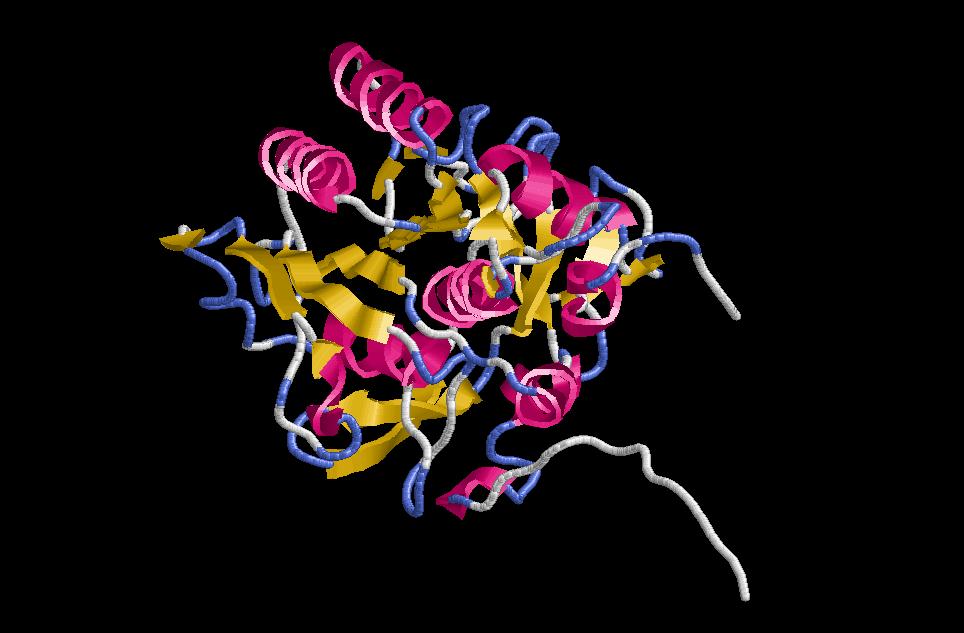
Corresponen als pdbs P11018.B99990001 i P11018.B99990002, respectivament.És d'esperar que siguin semblants entre ells i semblants també al model de clustalW. Trobem els mateixos problemes de loops que en l'altre cas i l'estructura secundària és aparenment igual. En detall podem trobar algunes diferències, però les diferències més significatives ens les trobarem quan utilitzem el programa prosa, en el càlcul de potencials estadístics.