Finalment, testaré el model optimitzat mitjançant Xam i Procheck.Comparació de models mitjançant Xam
Comparació de models mitjançant Xam
Primerament compararé el model original amb el model optimitzat mitjançant Xam pel càlcul de l'RMSD (veure pràctica 4.1). Els valors de RMSD obtinguts han estat (veure resultats complerts ):
1 2 
1 0.60 2 0.0 click to enlarge
Els valor de RMSD per l'alineament és petit, de 0.60, per tant indica que no hi ha grans diferències entre els models. La visualització de l'arxiu que en conté les coordenades també dóna la mateixa impressió, de manera que es pot arribar a la conclusió que Grumos no ha provocat canvis substancials en l'estructura de la proteïna a l'hora d'optimitzar, sinó que sobretot ha canviat l'orientació dels residus per tal de donar-los una configuració energètica més favorable.
Comparació de models mitjançant Procheck
L'anàlisi del sumari del model optimitzat (veure sumari) mostra que el nombre de mals contactes entre els àtoms del model ha passat a ser 0, mentre que abans era de 12 (veure sumari model 1.1), de manera que podem dir que el model ha millorat.
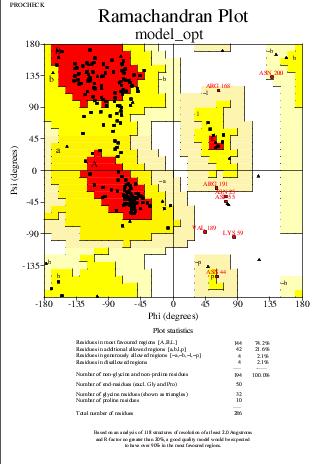
El mapa de Ramachandran, en canvi, mostra que hi ha només un 95.8% de residus en zones permeses (abans d'optimitzar eren un 97.8%), i en zones no permeses o permeses generosament s'ha incrementat fins al 4.2% (abans, 2.2%). Tot i això, la desaparició de residus experimentant mals contactes justifica la millora del model encara que la posició dels residus hagi empitjorat.
Download in .ps format Arxiu amb més definició. En Windows, obrir amb Adobe Photoshop. Comprimit amb WinZip. click to enlarge