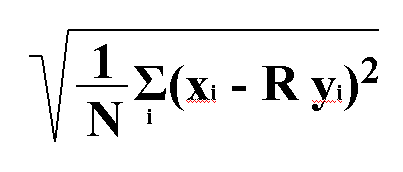En aquesta prŕctica realitzaré alineaments estructurals mitjançant el programa Xam, concretament per l'opció que fa referčncia al cŕlcul de l'RMSD (root mean square deviation).Primerament explicaré quč és l'RMSD i com es realitza l'execució del programa, i posteriorment efectuaré dos alineaments: un entre 4 hčlix α i l'altre entre dos plegaments globin-like de la família globin.
Cŕlcul de l'RMSD
Per realitzar l'alineament, el Xam realitza la torsió i translació de les molčcules a analitzar per tal que aquestes coincideixin i cerca residus comuns entre les dues cadenes:
El sumatori de la diferčncia entr x i y (on y es multiplica pel factor de rotació i translació):
ε |xi - R(yi)| (x i y vectorials) ha de ser el més petit possible, ja que a més petit, menys distŕncia hi haurŕ entre els residus comuns. A partir d'aquí es calcula el valor de RMSD, segons la fórmula:
Segons el valor de RMSD es pot valorar l'alineament:
VALORS DE RMSD Valor Alineament <2 A Bo 2-4 A Acceptable >4 A Dolent Aquests valors són aplicables per proteďnes. En pčptids de curt tamany es considera un bon alineament per sota els 0.5 A.
Per fer l'alineament estructural i el cŕlcul de l'RMSD, primerament executaré el programa:
/disc9/Superposition/xam/xam En aquest moment s'inicia Xam i et demana el nom del fitxer de sortida i dels fitxers d'entrada. Com que no tinc un fitxer amb el llistat de les estructures, n'introduiré el nom una a una:
Output file name : output1234_5 Input file list? or <cr> : Structure 1 or <cr> : helix1.pdb Structure 2 or <cr> : helix2.pdb Structure 3 or <cr> : helix3.pdb Structure 4 or <cr> : helix4.pdb Structure 5 or <cr> :Una vegada introduďdes les seqüčncies Xam obre els fitxers i en comprova la validesa:
Open file: helix1.pdb REMARK TITLE Total residues: 23 total atoms: 199 in structure: 1 Open file: helix2.pdb ATOM 304 N SER 37 -9.461 213.395 81.694 Total residues: 18 total atoms: 150 in structure: 2 Open file: helix3.pdb ATOM 1 N LYS 1 -30.968 220.204 69.410 Total residues: 14 total atoms: 115 in structure: 3 Open file: helix4.pdb ATOM 1 N ARG 1 -9.673 209.345 81.811 Total residues: 14 total atoms: 115 in structure: 4 Total molecules: 4 total groups: 1A continuació, et demana quina opció vols executar. En aquesta prŕctica realitzaré el cŕlcul de l'RMSD (opció 1):
1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML, 7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM, 9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 1El Xam mostra aleshores una sčrie d'opcions, de les quals agafarem les que s'executen per defecte. També demana un nom de fitxer on guardar la imatge de l'estructura superposada, perň en aquest moment no introduirem cap nom, ja que si no no mostraria el cŕlcul de l'RMSD:
Backbone atoms: N,CA,C,P,O5',C5',C4',C3',O3' BB atoms are listed above, if yes <cr> : Cyclopeptide? default is not, if OK <cr> : RMSD of 1st str. to the rest? default is not: Output file for superimposed struc.? or <cr> : Output file for mean structure? or <cr> :Finalment, demana que introdueixis els fragments a alinear si no corresponen als mateixos aminoŕcids en totes les estructures. En aquest cas s'analitza un sol fragment, perň si l'alineament de les estructures tingués gaps, aquestes s'haurien de fraccionar en diferents fragments:
Fragments for superposition, (default: all) first & last residue of frag. 1: 6 17 first & last residue of frag. 2: Selected residues for SC? filename or <cr>: Molecule 2 has different length! If the fragments for superimp. are different, give the ranges, otherwise <cr> first & last residue of frag. 1: 37 48 first & last residue of frag. 2: Molecule 3 has different length! If the fragments for superimp. are different, give the ranges, otherwise <cr> first & last residue of frag. 1: 1 12 first & last residue of frag. 2: Molecule 4 has different length! If the fragments for superimp. are different, give the ranges, otherwise <cr> first & last residue of frag. 1: 1 12 first & last residue of frag. 2:Una vegada introduďdes les posicions, el Xam fa el cŕlcul de l'RMSD i mostra de nou les opcions inicials, permetent realitzar un altre cŕlcul. En aquest punt, li tornaré a demanar que calculi l'RMSD per poder tenir una imatge visualitzable al RasMol (cal recordar que abans no he introduit cap nom en l'opció Output file for superimposed struc.):
Atom order not checked, because of different molecular lengthes or identifying atoms to be displayed! Atom order not checked, because of different molecular lengthes or identifying atoms to be displayed! Warning: no calculations for heavy atoms, its number not equal in molecule 2 1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML, 7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM, 9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 1Ara repetiré els passos anteriors (no mostrats), amb la diferčncia que sí que introduiré un nom de fitxer per guardar la imatge de la superposició:
Output file for superimposed struc.? or <cr>: superimp1234_5.pdbAl introduir aquest nom s'obren quatre noves opcions. Només no prendrem per defecte la primera:
Output fmt 1=DG,2=PDB,3=AMBER,4=bPDB,5=GSF,<cr>=DG 4 Key words (4 lettes, e.g., 5PTI) : A (all atoms) or S (superimposed)? <cr>=A Output for BB or Heavy superim.? B/H <cr>=BA partir d'aquest punt la resta de l'execució és igual que en el cas anterior. Quan torni a sortir el menú, ara escolliré l'opció 0: sortir.