Pràctica
6.2- Comparació de models
Pràctiques de Biologia Estructural-
Carlos Masdeu Ávila- Nia=16965
<< >>
1-Comparació
d'estructures secundàries
Ara ens fixarem en com són les estructures secundàries dels nostres
models i els evaluarem en funció d'aquestes. Utilitzarem dos programes:
Dssp,que fa un càlcul
(a partir de l'estructura) per trobar l'estructura secundària de la proteína,
i Psi-pred, que utilitza una base de dades per avaluar la seqüència
i oferir-nos així un percentatge de probabilitat.
1.1-Psi-pred
Donem la comanda
psipred clustal1.fa. Ell
realitza un psiblast, i en funció dels resultats, ens oferirà
un mapa d'estructura secundària o un altre.
Ens genera dos outputs,
on s'ens indica el tipus d'estructura secundària que ell preveu a cada
residu, així com la probabilitat associada.
clustal1.ss2
clustal1.horiz
(H vol dir helix, C vol dir unió, E vol dir làmina. Els nombres
corresponen a probabilitats. Hi ha un script anomenat psipred.pl que converteix
aquests aliniaments a format clustal, més entenedor: psipredpl.out)
1.2-DSSP
Dssp es crida
de la següent manera: dssp <input>
<output>
p.ex:
dssp
clustal1.pdb clustal1.dssp
L'output
de dssp, al provenir d'un input molt més complexe (pdb vs sequencia
FASTA), és també molt més complet i complexe. D'una
manera simil·lar a com ho feia psipred.pl, aliss es un script que, donant-li
un ..dssp, ens dòna la estructura secundària en format FASTA.
aliss.pl clustal1.dssp
> clustal1.2d
1.3-Comparant
estructures secundàries
A fi de comparar els outputs emesos per tots dos programes, crearem un fitxer
que contingui:
-la seqüència en fasta
-el output de dssp en fasta
-el output de psipred en fasta
Per fer-ho,
|
cat clustal1.2d
psipredpl.out > clustal1.2nd
(Concatenem
tots dos outputs en un sol fitxer)
(accesòriament
podem retallar la seqüència amb un editor gràfic, ja
que es trobarà repetida)
aconvertMod2.pl -in p -out c < clustal1.2nd > clustal1.2ncomp
(I
ho convertim a format clustal)
|
Així,
tenim a clustal1.2ncomp
els alineaments de secuència i estructura secundària del model
clustal1.
2-COMPARACIÓ
DE MODELS I SELECCIÓ DEL MILLOR MODEL
Ara que ja tenim
els models i coneixem les eines per comparar-los, passarem a comparar els diferents
models mitjançant cadascun dels programes que hem anat veient:
2.1-Procheck
2.2-Prosa
2.2b-Análisi amb procheck II
2.3-Superposició d'estructures
2.4-Psipred/dssp
2.1-Análisis
per procheck
2.1.1-RAMACHANDRAN
PLOTS
|
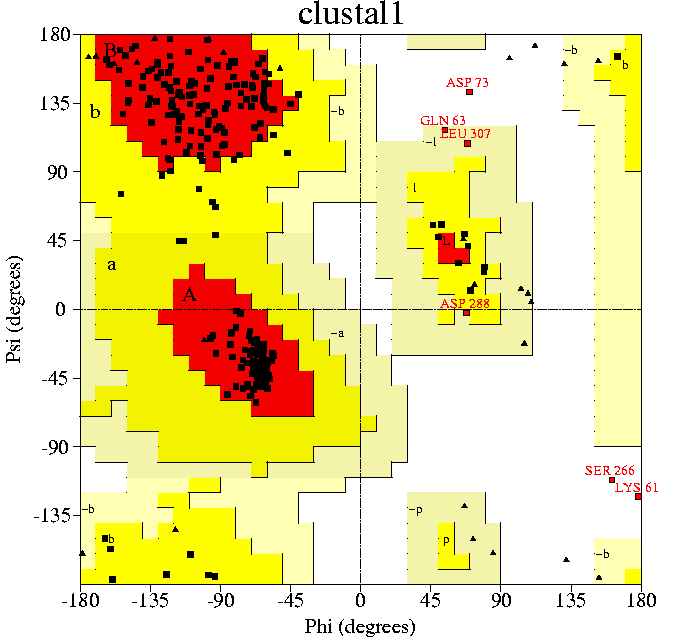
|
Al model clustal1 trobem que hi ha 3 residus (lys 61,asp73,ser266) que estàn
en posicions "disallowed" (no permeses) mentre n'hi ha 3 que estan
en regions rarament permeses. |
|
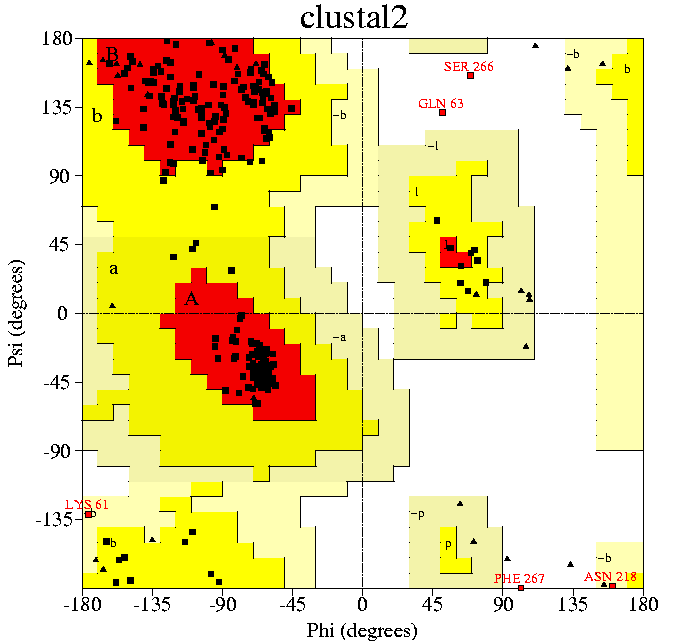
|
Aquest model (segon model generat amb l'alineament de seqüències)
té molt més bona pinta que l'anterior. Trobem que el nombre
de residus 'generosament' permesos és molt menor (només apareixen
2 al plot), i tenim tres residus que estan en posicions desfavorables (gln63,ser266,phe267) |
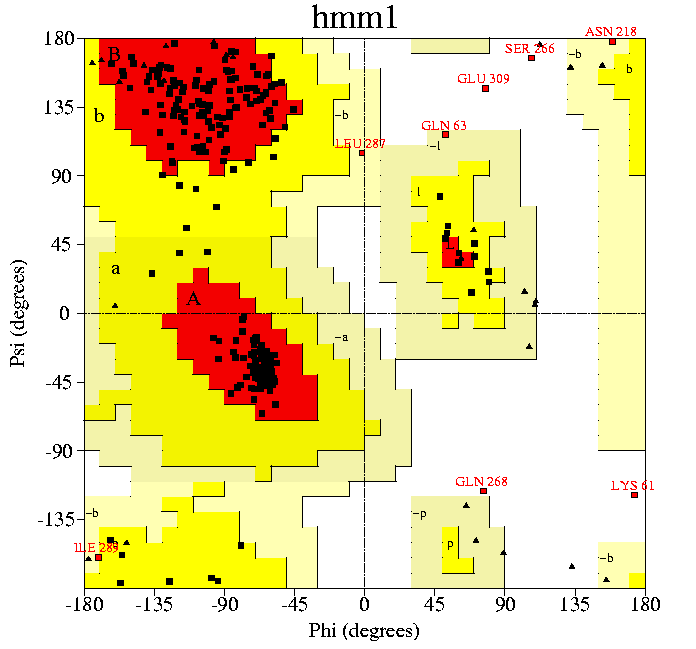 |
Tal com es veu el plot, hi ha molts residus que estàn a una zona
desfavorable de la meitat dreta, tant al quadrant superior com inferior.
Pel que fa als àtoms "generosament", en trobem bastants
repartits pel plot. |
|
hmm2
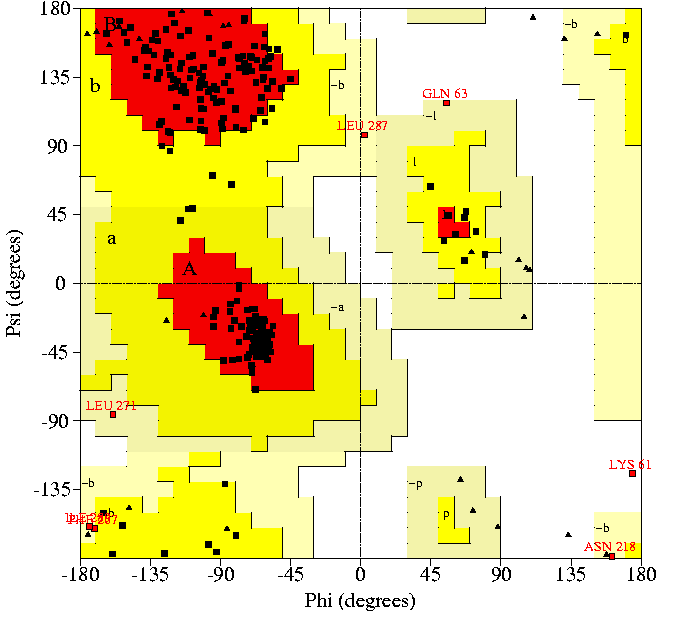
|
Aquest model està millorat respecte a l'altre realitzat amb l'alineament
de hmm. Tal com es veu al plot, les úniques posicions que ens criden
l'atenció són àtoms que estàn en posicions permeses
"generosament",tret de la lys 61 que, tal com passava a l'altre
model, queda en la zona "disallowed". |
2.1.2-Summaries
|
Model
|
core
|
allow
|
gener
|
disall
|
Bad
contacts
|
|
|
86.7%
|
11.1%
|
1.1%
|
1.1%
|
8
|
|
|
88.2%
|
10.0%
|
0.7%
|
1.1%
|
9
|
|
|
87.1%
|
10.0%
|
1.5%
|
1.5%
|
7
|
|
|
87.5%
|
10.0%
|
2.2%
|
0.4%
|
9
|
Tal
com podem veure als plots i al summary, tots quatre models presenten residus
amb característiques enenergèticament desfavorables i, si ens
fixem bé, veurem que la suma de 'disallowed' i 'generously allowed' (és
a dir, el conjunt de residus que són incorrectes o bé correctes
amb un dintell d'error massa alt) és bastant simil·lar als quatre.
(clu1:2.2%, clu2:1.8%,hmm1:3%, hmm2:2.6%).
No obstant, no és fàcil
decantarse per cap dels quatre models, ja que aquell que té un més
alt percentatge de residus en el 'core' i menor en disallowed (hmm2) és
precissament el mateix que té el major nombre de bad contacts, i el que
té el menor nombre de bad contacts és el que té el major
percentatge de disallowed.
2-Anàlisi
per prosaII
Passem, per tant, a analitzar
els nostres models amb prosa:
-Carreguem tots quatre
pdb's
-Demanem que ens faci un analisis d'energia (analyse
energy *)
-Assignem els colors:
color * hmm1 green
color * hmm2 blue
color * clu1 yellow
color * clu2 red
-Canviem l'amplada de
la finestra lliscant: winsize * 50
i grafiquem:
plot
pscolor = 1
export my plot c12h12.ps
Anem a veure
els resultats...

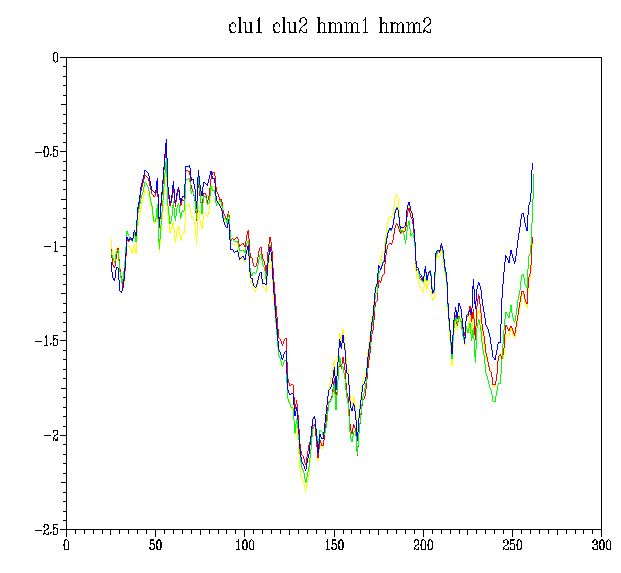
Tal com esperàvem, tots quatre
models són molt semblants, ja que el nombre de seqüències
que vam utilitzar per crear-los era baix. No obstant, es veu a simple vista
que són prou estables energèticament, tot i que a partir de l'aa
275 aproximadament, totes les gràfiques d'energia comencen a pujar i
a tornar-se energèticament desfavorables.
Obrirem els
models amb RasMol i intentarem veure a què es degut...
Tal com es veu a la captura
de RasMol, totes les seqüències tenen uns segments llargs i sense
estructura secundària que són, de ben segur, resultat de les regions
terminals de P11018 que no trobaven homologia. Per arreglar aquest problema,
veurem a partir de quin aa comença el problema, i 'retallarem' els models
treient aquestes llargues cues del fitxer.
D'acord amb el que veiem
a prosa i a RasMol, sembla que a partir de l'aa 300 és cuan comencen
els problemes.Analitzant els alineaments, veiem que les regions on P11018 no
trobava homologia eren els aa 1-18 i 305-320
Retallant
els models
Per fer-ho, obrim
el fitxer en mode texte (emacs), i retallem aquells aminoàcids dels extrems
que no s'alineaven. Amb els del principi, només cal 'retallar', però
a l'hora de treure els del final, cal tenir en compte que el darrer àtom
ha de ser un OXT, i ha de tenir unes coordenades coherents amb el anterior àtom.
La manera de solucionar-ho es la següent:
Suposem que volem retallar tots aquells residus més enllà del
304. Al pdb, trobem el següent:
ATOM 2254 N PRO 304 13.233
62.471 24.141 1.00160.46 1SG2255
ATOM 2255 CA PRO 304 13.894 61.675 23.153 1.00160.46 1SG2256
ATOM 2256 CD PRO 304 13.670 61.945 25.423 1.00160.46 1SG2257
ATOM 2257 CB PRO 304 13.854 60.250 23.681 1.00160.46 1SG2258
ATOM 2258 CG PRO 304 14.137 60.494 25.173 1.00160.46 1SG2259
ATOM 2259 C PRO 304 13.553 61.884 21.718 1.00160.46 1SG2260
ATOM 2260 O PRO 304 14.419 61.626 20.884 1.00160.46
1SG2261
ATOM 2261 N ASP 305 12.335 62.335 21.385 1.00 84.18 1SG2262
ATOM 2262 CA ASP 305 12.052 62.563 19.998 1.00 84.18 1SG2263
ATOM 2263 CB ASP 305 10.586 62.907 19.690 1.00 84.18 1SG2264
ATOM 2264 CG ASP 305 9.790 61.612 19.668 1.00 84.18 1SG2265
ATOM 2265 OD1 ASP 305 10.183 60.661 20.394 1.00 84.18 1SG2266
ATOM 2266 OD2 ASP 305 8.779 61.558 18.918 1.00 84.18 1SG2267
ATOM 2267 C ASP 305 12.884 63.697 19.507 1.00 84.18 1SG2268
Per tancar la proteína
al 304, borrem tots els residus posteriors, excepte l'atom inmediatament posterior
al O de la PRO 304 (en aquest cas, seria el atom 2261). Substituim N per OXT
(és a dir, ara ja no hi ha enllaç peptídic, demanera que
en lloc de amino hi haurà l'oxigen del carboxi terminal), i les columnes
corresponents a dades del residu 305 les substituim com si fossin del 304, però
deixant les coordenades intactes. A la següent linia, afegim un TER o un
END per indicar que ja s'ha acabat la proteína, i ja hem tret la part
que ens molestava. Quedaria de la següent manera:
ATOM 2254 N PRO 304 13.233
62.471 24.141 1.00160.46 1SG2255
ATOM 2255 CA PRO 304 13.894 61.675 23.153 1.00160.46 1SG2256
ATOM 2256 CD PRO 304 13.670 61.945 25.423 1.00160.46 1SG2257
ATOM 2257 CB PRO 304 13.854 60.250 23.681 1.00160.46 1SG2258
ATOM 2258 CG PRO 304 14.137 60.494 25.173 1.00160.46 1SG2259
ATOM 2259 C PRO 304 13.553 61.884 21.718 1.00160.46 1SG2260
ATOM 2260 O PRO 304 14.419 61.626 20.884 1.00160.46 1SG2261
ATOM 2261 OXT PRO 304 12.335 62.335 21.385 1.00 84.18
1SG2262
END
Veiem ara com queda a prosaII...
(abans)
(ara)
Tal com es veu comparant
totes dues representacions d'energia, retallant els extrems 'no aliniats' de
la proteína, hem aconseguit millorar molt el perfil energètic
de les estructures (si més no, per els extrems)
En
quant a l'avaluació de les estructures, podem concloure que tots quatre
models tenen unes gràfiques d'energia prou simil·lars i que cap
d'ells arriba a valors positius, de manera que per aquesta banda gairebé
podríem acceptar-los tots quatre com mitjanament correctes.
2b-ANALISI
PER PROCHECK II
(com hem retocat les estructures al veure les gràfiques a prosaII, tornarem
a fer un anàlisi amb procheck de tots quatre models, a veure si milloren
una mica els plots...)
(Els plots no s'adjunten ja que són extremadament semblants als anteriors.
Només desapareixen aquells residus que estaven 'mal ubicats' i que pertanyien
a un dels extrems tallats. Els summaries, en canvi, sí que han variat
(han variat els percentatges. Els nous són els següents: peloscortos.sum
Els resultats actuals són:
|
Model
|
core
|
allow
|
gener
|
disall
|
Bad
contacts
|
|
|
86.3%
|
11.6%
|
0.8%
|
1.2%
|
6
|
|
|
87.6%
|
10.4%
|
0.8%
|
1.2%
|
8
|
|
|
85.9%
|
11.2%
|
1.7%
|
1.2%
|
7
|
|
|
86.7%
|
10.4%
|
2.5%
|
0.4%
|
7
|
3-SUPERPOSICIO
D'ESTRUCTURES
Utilitzarem STAMP per fer un alineament estructural dels cadascun dels nostres
models amb les estructures a partir de les quals els vam crear.
Primer passem PDBtoSplitChain.pl
a tots els models.
Creem els fitxers .domain perquè STAMP pugui funcionar, i cridem STAMP:
stamp -l clustal1.domain
-rough -n 2 -prefix clustal1
stamp -l clustal2.domain -rough -n 2 -prefix clustal2
stamp -l hmm1.domain -rough -n 2 -prefix hmm1
stamp -l hmm2.domain -rough -n 2 -prefix hmm2
Els valors de RMS i score
d'alineament de cadascun dels models, (ordenats per ordre creixent de RMS),
són:
hmm1:
0.264700 Sc = 9.294363
hmm2: 0.290252 Sc = 9.273805
clu1: 0.337572 Sc = 9.213210
clu2: 0.398268 Sc = 9.280082
Tal com es veu, tots quatre
models presenten un alineament prou bo amb la resta d'estructures de la família
(molt inferior a 1), de manera que aquí encara no en podem descartar
cap d'ells.
4-Comparació
per estructura secundària
L'estratègia que
seguirem és la següent:
-Ja que estem tractant amb estructures, i no seqüències -tots
els models tenen la mateixa seqüència,després de tot!-,
obtindrem, amb dssp, les estructures
secundàries de les 4 proteínes amb les que vam construir els models.
-Llavors, les mostrarem en un alíniament (tal com vam fer al principi
d'aquesta pràctica) per intentar veure quina s'ajusta més al patró
de la família.
dssp clu1.pdb clu1.dssp
dssp clu2.pdb clu2.dssp
dssp hm1.pdb hmm1.dssp
dssp hm2.pdb hmm2.dssp
aliss.pl clu1.dssp > clu1.2nd
aliss.pl clu2.dssp > clu2.2nd
aliss.pl hmm1.dssp > hmm1.2nd
aliss.pl hmm2.dssp > hmm2.2nd
L'alineament d'estructures
secundàries, tal com es veu quan les aliniem totes, surt molt semblant
a tots quatre models -de nou, el motiu és haver escollit tan poques seqüències
per construir els models- , però tot i així ja hi ha alguna diferència
que ens permet veure, per exemple, que clustal1 és el que ha conservat
més salguns dominis que els altres models han perdut.
5-La tria
del model
Com s'ha anat
veient, tots quatre models semblen prou aptes, i les diferències dels
diferents anàlisis no van -ni molt menys- en direcció a afavorir
cap d'ells. No obstant, clustal1 ha mostrat uns resultats mitjos millors a la
resta (especialment a ProsaII i a Procheck).
El valor obtingut a la RMS de STAMP tampoc és dolent (0.33), així
que clustal1.pdb serà el model que triem per a fer la següent
i darrera pràctica.