Pràctica
7- Optimització d'estructures: GRUMOS
Pràctiques
de Biologia Estructural- Carlos Masdeu Ávila- Nia=16965
<<
1-Introducció
GROMOS
és un software d'optimització d'estructures mitjançant
"optimització geomètrica", en el que
s'intenten assolir mínims energètics intentant reduir
paràmetres com forces de torsió en els angles dels
àtoms ,etc.. presents al nostre model, derivades de l'intent
de construir una estructura quimèrica a partir de petites
parts amb les que, seqüèncialment, hi havia homologia.
Nosaltres
utilitzarem GRUMOS, que és una versió més còmoda
d'utilitzar, a fi d'optimitzar el model que vam seleccionar d'entre
tots els generats.
És
important que el nostre .pdb input compleixi una sèrie de
requisits per al correcte funcionament amb GRUMOS. Per això,
primer li passarem un script que arregla els pdb per a grumos,
arrangeG.pl. Una altra
cosa important de cara a la compatibilitat amb GRUMOS és que
el nostre pdb acabi amb una linea en la que només apareix TER.
Preparat
ja el pdb, l'optimitzarem mitjançant GRUMOS...
2-L'interfície
GRUMOS
-Primer
ens demanarà el fitxer d'entrada i el directori per a crear
l'output.
-Triem
Input>Topology>simple system
-Ens
demana un títol
-Després
ens preguntarà si volem canviar el nom d'algun aminoàcid
de la nostra seqüència. Això serveix per declarar
ponts disulfur, per exemple, en els què les cisteïnes que
intervenen en el pont passen a dir-se CYSH
o CYS1, CYS2. En el
nostre cas, he mirat, mitjançant RasMol, si havia dos
cisteines prou properes com per formar un pont disulfur, però
les dos cisteínes de la estructura estaven massa lluny com per
formar un disulfur. Així, no declararé cap pont
disulfur.
-Triem
Input>Topology>simple system
-Em
pregunta pel solvent,i trio la opció per defecte, <1>
Non inertial solvent, NIS model
-Em
pregunta per la ionització, i trio la que hi hauria a pH=7
-Llavors
correm el procés de Topology (single system)
(després
de que GRUMOS donés molts problemes amb el model, he acabat
adonant-me que hi havia un error a una de les seqüències
utilitzades per a fer el model, i a les isoleucines hi havia un atom
que ell deia CD1 quan grumos esperava un CD... al no trobar-lo,creia
que li faltava un àtom, i no podia continuar amb el procés.
He editat el pdb des de kwrite, substituint tots els àtoms CD1
de les ILE per CD; després de fer-ho, sí que accepta el
model, de manera que puc passar al següent pas)
-Ens dirigim
ara a Energy Optimization. Primer creem l'input:
-Utilitzarem
el procediment d'Steepest Descent, que busca la manera més ràpida
d'arribar als mínims energètics
-1000 pasos
per cada execució, 10 optimitzacions
-Respecte els
valors de Lambda, agafem les opcions per defecte
-No usem SHAKE
-Interaccions
per parelles de GRUPS i no d'àtoms
-Value of RCUTP
< 0.8 nm> : 0.8
-Value of RSWI2
<10.0 nm> : 99999 (augmentem el valor perquè el radi sigui tan alt
que no ho calculi)
-Value of RCUI2
<10.0 nm> : 99999 (idem)
-Value of RCUTL
< 1.3 nm> : 1.3
-Sequence radius
to calculate the interaction: 999999
-No periodicity,
no position restraining, No distance restraining
Un cop creat
l'input, ja podem correr el procés:
RUN a process>ENERGY
OPTIMITZATION>Single system
El programa
queda llavors en background, de manera que si sortim i anem al directori que
hem definit per a l'output, veurem com GRUMOS va fent els processos.
3-L'output
de GRUMOS
-Dins de la
carpeta generada per GRUMOS, trobem una sèrie numerada d'arxius .lis,
que són l'informe d'energia a cada iteració. Aquí s'adjunten
3 per poder veure l'evolució:
outclu_emnum001.lis
outclu_emnum006.lis outclu_emnum010.lis
(L'evolució
del procés es pot veure si anem a la part 3, ATOM COORDINATES, on apareixeran
quadres com aquest( s'ha marcat en vermell el nombre que correspon al nombre
de Step d'aquella iteració i en groc l'energia potencial total de la
proteína en aquest pas):
STEP
NIP1 NIS1 NIP2 NIS2 NIP3 NIS3 NITI STEP-SIZE RMS-F-FREE RMS-F-CONS
E-POT-TOT E-BOND-H E-BOND E-ANGLE-H E-ANGLE E-IM-DIH-H E-IM-DIH E-DIH-H
E-DIHEDRAL
E-EL-G1-G1 E-EL-G1-G2 E-EL-G2-G2 E-EL-G1-G3 E-EL-G2-G3 E-EL-G3-G3 E-EL-G1-G4
E-EL-G2-G4 E-EL-G3-G4 E-EL-G4-G4
E-LJ-G1-G1 E-LJ-G1-G2 E-LJ-G2-G2 E-LJ-G1-G3 E-LJ-G2-G3 E-LJ-G3-G3 E-LJ-G1-G4
E-LJ-G2-G4 E-LJ-G3-G4 E-LJ-G4-G4
E-DIS-R. E-POS-R. E-DIH-R.
100 1 0 0 0 0 0 0 0.1294E-02 0.8343E+01
0.8343E+01
-0.16502E+05 0.1750E+02 0.1894E+03
0.1892E+03 0.1309E+04 0.1654E+03 0.3390E+03 0.4558E+02 0.1101E+04
-0.8677E+04 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00
0.0000E+00 0.0000E+00 0.0000E+00
-0.1118E+05 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00
0.0000E+00 0.0000E+00 0.0000E+00
0.0000E+00 0.0000E+00 0.6815-269
|
(Tal com
es veu, si es miren el primer pas d'outclu_emnum001.lis i el darrer de outclu_emnum010.lis,
l'energia potencial total del sistema passa de 0.12903E+05 a -0.16727E+05)
-GRUMOS també
ens genera un altre output, que es troba a la carpeta coordinates
dins del directori que hem marcat com a ouput.
-Aquí
trobarem també una sèrie d'arxius numerats, però en aquest
cas tenen l'extensió .gsf. Corresponen
a les coordenades de l'estructura després de cada pas d'optimització,
i si els mirem, veurem com el format, tot i que s'assembla al format pdb, no
acaba de ser exactament igual (falten les paraules clau ATOM, la divisió
en columnes no es igual, etc...)
-El model corresponent
a la darrera optimització és copiat, a més del .gsf, amb
un altre nom, cluxvmd0000.gsf,
però el format continua
essent el mateix.
4-Superposant
els models optimitzats (XAM)
-A fi de veure
l'evolució que ha anat fent el model al llarg de l'steepest descent (i
convertir de pas els .gsf en un format .pdb visible amb RasMol), superposem
alguns dels models amb XAM. (veure prac4.1)
 |
Tal
com es veu, la superposició de tres dels models (corresponents als
pasos 1,5 i 10) sembla no revelar gaire diferències entre ells, quan
es representa en forma 'cartoon' amb els colors per estructures. |
 |
No
obstant, si canviem el mode de vista a Wireframe i pintem cada model d'un
color, veurem com hi ha petites variacions a gairebé tots els angles
entre àtoms (veure requadres grocs).
Si
tenim en compte la gran quantitat d'angles que formen tota l'estructura,
podem fer-nos una idea de la millora energètica que pot suposar
el sumatori de la contribució energètica de cadascun d'aquests
petits angles.
|
-Fet això,
ja podem extreure el nostre model del fitxer de superposició. Basta amb
trobar la linea on comenci el darrer dels models, i copiar-ho a un altre arxiu
amb el nou nom.
-Un cop hem
separat el model optimitzat, podem passar doncs a avaluar-lo.
5-Avaluació
del model optimitzat
-Tal com vam
fer a la pràctica anterior, utilitzarem els següents programes per
avaluar el nostre model:
5.1-Procheck
5.2-Prosa
5.3-Superposició d'estructures
5.4-Psipred/dssp
5.1-Avaluació
del model amb Procheck
Ramachandran
Plot:
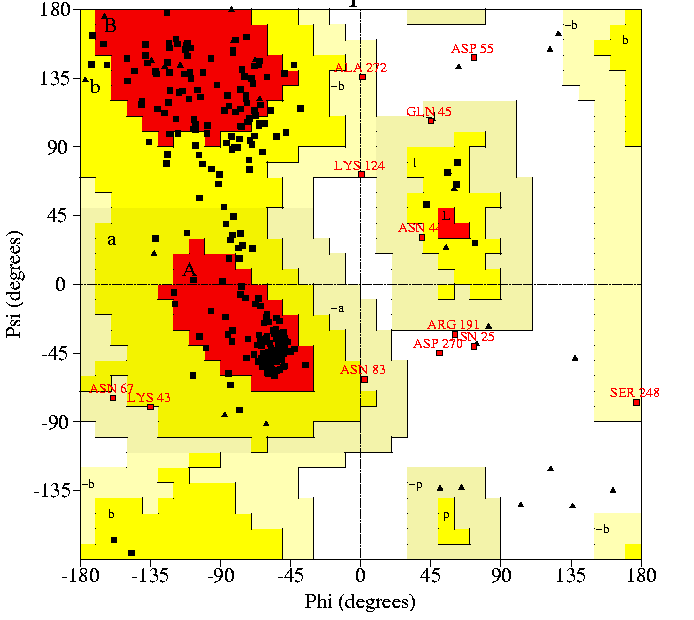 |
El
Ramachandran Plot sembla haver empitjorat: apareixen varis residus a zones
'disallowed', cosa que abans no ocorria, i ha augmentat el nombre de residus
situats a les zones 'generously allowed' |
Summary:
modeloptsuelto.sum
Comparant
amb abans de l'optimització,
|
Model
|
core
|
allow
|
gener
|
disall
|
Bad
contacts:
|
|
Clustal1.pdb
|
86.7%
|
11.1%
|
1.1%
|
1.1%
|
8
|
|
Clustal1
Optimizado
|
73.0%
|
22.0%
|
3.3%
|
1.7%
|
0
|
Veiem
que hi ha hagut una redistribució de residus en les diferents zones del
plot, amb un augment de les zones energèticament més desafavorides
en detriment del core. La zona de residus permesos, no obstant, ha augmentat
en ocurrències. El canvi més notori és el nombre de bad
contacts, que ha estat reduït a zero, fet que millora molt la versemblança
del model.
5.2-Avaluació
amb ProsaII
Per
a realitzar l'avaluació amb prosaII, graficarem l'energia del model abans
i després de l'optimització de GRUMOS.
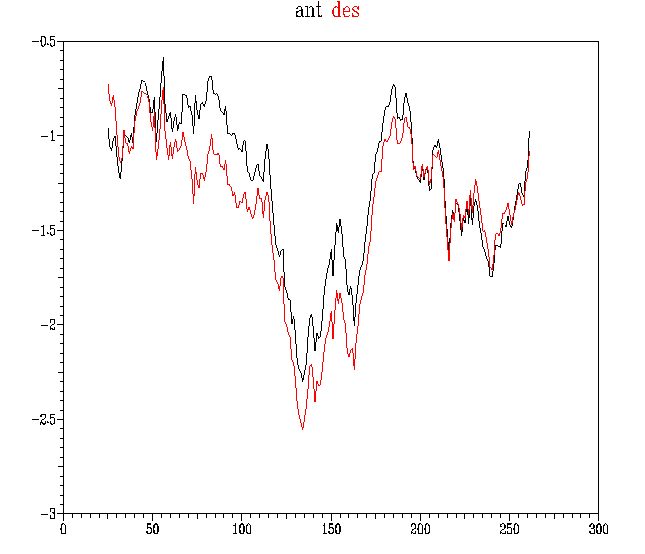 |
Afortunadament,
l'optimització ha millorat molt el perfil energètic del nostre
model, especialment en la vall central.
L'extrem NH3 (esquerra de la gràfica) ha vist augmentada lleugerament
la seva energia, però continua sent negativa, de manera que no ens
preocuparem gaire per aquest fet. |
5.3-Superposició
d'estructures
De
nou, utilitzarem STAMP per superposar l'estructura amb les homòlogues
per veure si ha variat gaire l'alineament estructural amb l'optimització.
L'output
ens indica que el RMS de la superposició del nostre model amb la resta
és de 0.61 (bastant més alt que el valor que donava el model abans
de passar per grumos, veure resultats de
superposició de la prac.6.2), la qual cosa vol dir que, estructuralment,
a l'optimitzar energèticament la proteïna ens hem allunyat del 'canon'
estructural definit per les 4 proteínes a partir de les quals vam construir
el model.
No
ens ha d'extranyar aquest fet, no obstant, ja que al haver-hi tanta petita reordenació
espacial, com vam veure a l'analitzar l'evolució del model al llarg de
l'optimització, el resultat final és normal que disti molt estructuralment
de com era a l'inici.
Transformem
l'output de la superposició en pdb, a fi de veure si aquestes diferències
estructurals són només deguts a moltes petites variacions en els
angles entre àtoms, o bé hi ha alguna cosa més gran i visible:
 |
Ja
amb la representació en mode 'Cartoon' es veu com el nostre model
optimitzat presenta variacions respecte a la resta d'estructures. En la
captura s'han senyalat quatre coils que segueixen una trajectoria completament
diferent a la que segueixen els homòlegs (la qual és, en tots
tres casos, bastant conservada). |
|
|
|
 |
En
la representació en mode wireframe veiem com moltes de les cadenes
laterals dels residus protueixen cap a l'exterior. Això és
degut segurament a què a l'optimització, a fi d'evitar 'bad
contacts', es deuen haver mogut aquestes cadenes laterals cap a aquells
llocs on tenien menys impediments estèrics (és a dir, en molts
dels casos, cap a fora) |
|
|
|
|
|
Aquesta
captura vé d'un aliniament addicional realitzat afegint la seqüencia
no optimitzada. El motiu era comprovar si abans de l'optimització
protuïen tant les cadenes laterals cap enfora. Tal com es veu a la
imatge, tots dos models (abans de la optimització
i després de la optimització) guarden
un patró simil·lar en aquest aspecte, de manera que no podem
explicar les diferències de RMS només amb aquest fet, sinò
que hem de tenir en compte que és el sumatori de molts petits canvis
el que ha acabat conformant el augment de RMS. |
5.4-Estructura
secundària
Utilitzarem
DSSP per obtenir la seqüència secundària del model en format
FASTA, i després l'aliniarem amb els altres de la família i amb
clustal1.dssp (model triat, abans d'optimitzar) , a fi de valorar el
canvi respecte a sí mateix i a la família de proteínes.
Un cop tenim l'output de dssp per al nostre model,
l'aliniem amb la resta, i obtenim opt.aln:
 |
He
intentat marcar amb colors les zones en les què el model optimitzat
suposa alguna millora clara respecte al no optimitzat(verd) i les què,
pel contrari, ha allunyat encara més de l'estructura de la família.
Tal
com vam veure amb la superposició estructural, l'optimització
també ha allunyat en aquest aspecte el model de les estructures
a partir de les quals va ser creat
|
Conclusions
respecte l'optimització
L'optimització
ha fet més estable el nostre model, però a expenses d'allunyar-lo
del patró estructural que marcaven els homòlegs a partir del quals
vam construir-lo. Això es deu sobretot al fet què hi havia massa
residus en "Bad Contact" o bé amb angles que no els feien ser
'allowed'. En conseqüència, la optimització energètica
ha estat més pronunciada, i s'han hagut de variar molt més les
estructures a fi de poder aconseguir uns mínims energètics, i
ha estat en aquest procés que les estructures s'han vist afectades.
<<
