 MÁS DIFÍCIL TODAVÍA...
MÁS DIFÍCIL TODAVÍA...
MÁS DIFÍCIL TODAVÍA...
MÁS DIFÍCIL TODAVÍA...
Bien, hasta ahora hemos construído un modelo de Floripondio que
tenía sus extremos mal modelados y que interferían con el
plegamiento del resto de la proteína, y viendo que había
un amiguete demasiado largo, lo hemos cortado para ver si esos extremos
mejoraban. Lo cierto es que han mejorado algo, pero no lo suficiente (nosotras
siempre queremos más). Por tanto, lo que haremos ahora será
cortar los extremos de floripondio (no lloréis, que la amputación
es pequeña y para mejor) y ver si el plegamiento del resto de la
molécula mejora o no.
Ya sabeis que esto de utilizar las tijeras es un poco peligroso, así que para no pasrnos cortaremos sólo aquellos residuos de la secuencia que no se alinean con el resto de templates. Crearemos dos modelos más:
![]() Modex:
en el que cortamos en base al alineamiento de secuencia.
Modex:
en el que cortamos en base al alineamiento de secuencia.
![]() Modcua: en el que cortaremos en base al alineamiento
de estructuras.
Modcua: en el que cortaremos en base al alineamiento
de estructuras.
![]() Modex,
o cortando sobre el papel:
Modex,
o cortando sobre el papel:
Para ver por dónde había que cortar la secuencia de floripondio, supusimos que los residuos que no se alineaban correctamente con el resto de templates eran los mismas que formaban las "colas" que no pudimos modelar. Por tanto, cortaremos a partir del punto en que el alineamiento modcut.ali(contiene los amiguetes, con 1emsA cortado, junto a floripondio y alineados según la matriz oculta de markov) empieza a ser malo.
En el link del alineamiento anterior, hemos señalado en rojo aquellos residuos que forman el centro activo de la proteína, y en verde los residuos que eliminamos para hacer el modelo.
Tras
una divertida sesión de manualidades, obtuvimos las nuevas secuencias
de las proteínas que las pasamos por los dos filtros (PDBtosplitchain
y arrangeG) y los reunimos en una pequeña fiesta (totex.fa)
para poder hacer un alineamiento de secuencia gracias al siempre fiel Clustalw
que guardamos en el archivo totex.aln.
Otra
vez los superpusimos con el STAMP para obtener la matriz oculta de Markov
(modex.hmm).
Ésta es una imagen de la fiesta:
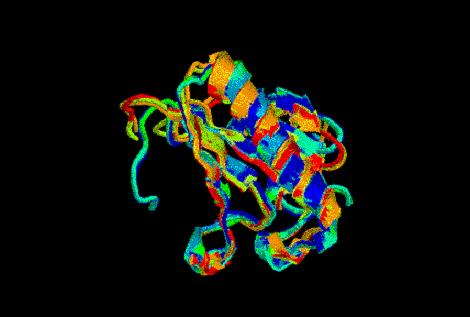
Una vez llegados a este punto, los alineamos con el perfil de markov (modex.ali)
para darle forma a nuestro pequeñín, y con la generosidad
del Modeller, obtuvimos las 2 estructuras de floripondio sin sus extremos
más conflictivos:
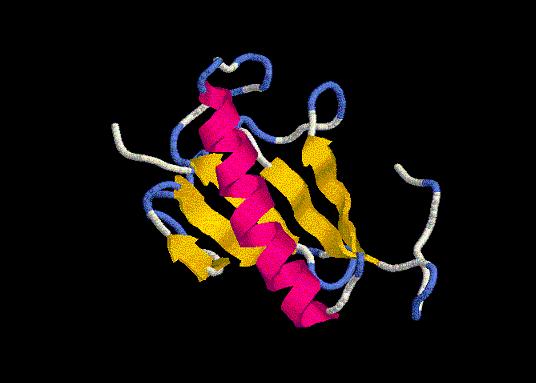

floriexA.pdb
floriexB.pdb
Oh, gran sorpresa! (y decepción, para que negarlo) con nuestro afán tijeretero eliminamos la última hélice. Por tanto, tuvimos que construir otro modelo, el modcua, en el que en lugar de cortar según el alineamiento de secuencia, lo hicimos según el alineamiento estructural.
Aún así, optimizamos el modelo y le hicimos el análisis
energético. Los resultados son los siguientes. Decidimos optimizar
el modelo floriexA.pdb, ya que sólo tenía 3 "bad contacts",
mientras que el modelo floriexB.pdb tenía 5.
<<< P R O C H E C K S U M M A R Y >>>
florexA.pdb 2.8 116 residues
* Ramachandran plot: 93.2% core 3.9% allow 1.9% gener 1.0% disall
All Ramachandrans:
3 labelled residues (out of 114)
* Chi1-chi2 plots:
3 labelled residues (out of 71)
Main-chain params:
6 better 0 inside
0 worse
Side-chain params:
5 better 0 inside
0 worse
Residue properties: Max.deviation:
4.0
Bad contacts: 2
Bond len/angle: 3.7 Morris et al class:
1 1 2
1 cis-peptides
G-factors
Dihedrals: -0.01 Covalent: -0.20 Overall:
-0.08
M/c bond lengths: 98.6% within
limits 1.4% highlighted
M/c bond angles: 93.0%
within limits 7.0% highlighted
Planar groups:
100.0% within limits 0.0% highlighted
<<< P R O C H E C K S U M M A R Y >>>
florexB.pdb 2.8 116 residues
Ramachandran plot: 88.3% core 10.7% allow 1.0% gener 0.0% disall
*All Ramachandrans:
5 labelled residues (out of 114)
+ Chi1-chi2 plots:
1 labelled residues (out of 71)
Main-chain params:
6 better 0 inside
0 worse
Side-chain params:
5 better 0 inside
0 worse
* Residue properties: Max.deviation:
2.3
Bad contacts: 5
*
Bond len/angle: 6.5 Morris et al class:
1 1 2
1 cis-peptides
G-factors
Dihedrals: -0.03 Covalent: -0.30 Overall:
-0.13
M/c bond lengths: 98.1% within
limits 1.9% highlighted
M/c bond angles: 92.5%
within limits 7.5% highlighted
Planar groups:
100.0% within limits 0.0% highlighted
Una vez optimizado el modelo, vimos que habíamos reducido a cero
el número de bad contacts, y que la energía era menor, pero
claro, seguía faltando la hélice. (Glups!)
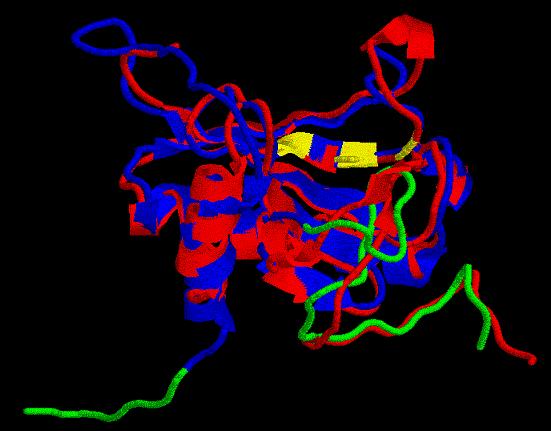
 Modcua: no nos pasemos cortando...
Modcua: no nos pasemos cortando...
Ya llegamos al final de nuestra aventura estructural:
Para hacer el recorte de las secuencias, hicimos una superposición
con el progrma STAMP de nuestro mejor modelo hasta el momento y nuestro
mejor template: 1emsA (con la secuencia cortada). Obtuvimos las coordenadas
de ambas proteínas (protein.pdb), y vimos
que los fragmentos que debíamos cortar eran de la metionina 1 a
la alanina 31 por el extremo N-terminal y de la alanina 153 a la
160 por el C-terminal.
En el fichero floricutAoptim.pdb eliminamos los 31 primer residuos y los 8 últimos residuos, lo pasamos por los 2 flitros de rigor y obtenemos el fichero que contiene las coordenadas (floricua.pdb) y el que contiene la secuencia (floricua.fa).
Añadimos la secuencia fasta a un fichero que contiene las secuencias del resto de amiguetes (totcua.fa) y lo alineamos con la matriz oculta de Markov modcut.hmm (es la misma que en el modelo modcut, ya que esta vez no hemos modificado nada de los templates), obteniendo así el alineamiento cua.ali. Este alineamiento se utilizó para modelar la secuencia floricua.fa con el Modeller, para conseguir los modelos modcuaA.pdb y modcuaB.pdb. (una pista: éstos serán los modelos definitivos)
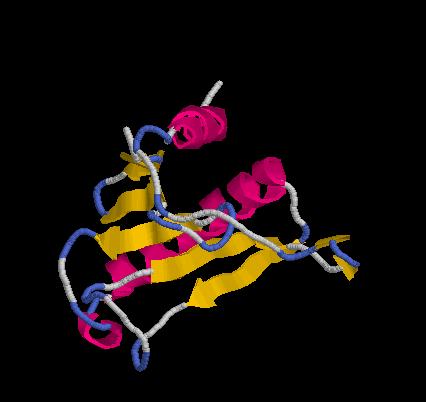
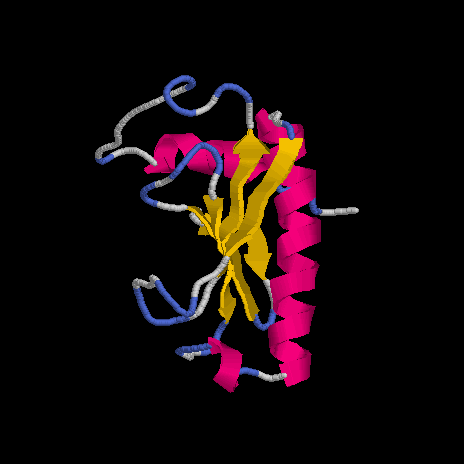
floricutA.pdb
floricutB.pdb
Otra vez aflora nuestro espíritu competitivo: debemos elegir entre
el modelo A y el B.
Pasamos
dichos modelos por el Procheck. Marcador: 6 bad contacts para el B y 3
para el A. La victoria del A es clara.
<<< P R O C H E C K S U M M A R Y >>>
floricuaA.pdb 2.8 121 residues
* Ramachandran plot: 89.8% core 8.3% allow 0.0% gener 1.9% disall
* All Ramachandrans:
5 labelled residues (out of 119)
Chi1-chi2 plots:
1 labelled residues (out of 77)
Main-chain params:
6 better 0 inside
0 worse
Side-chain params:
5 better 0 inside
0 worse
* Residue properties: Max.deviation:
4.0 Bad
contacts: 3
*
Bond len/angle: 6.3 Morris et al class:
1 1 2
1 cis-peptides
G-factors
Dihedrals: -0.04 Covalent: -0.25 Overall:
-0.12
M/c bond lengths: 99.2% within
limits 0.8% highlighted
M/c bond angles: 91.6%
within limits 8.4% highlighted
Planar groups:
100.0% within limits 0.0% highlighted
<<< P R O C H E C K S U M M A R Y >>>
floricuaB.pdb 2.8 121 residues
* Ramachandran plot: 88.9% core 9.3% allow 0.0% gener 1.9% disall
* All Ramachandrans:
4 labelled residues (out of 119)
Chi1-chi2 plots:
2 labelled residues (out of 77)
Main-chain params:
6 better 0 inside
0 worse
Side-chain params:
5 better 0 inside
0 worse
Residue properties: Max.deviation:
4.0 Bad
contacts: 6
*
Bond len/angle: 6.3 Morris et al class:
1 1 2
G-factors Dihedrals: 0.03 Covalent: -0.25 Overall: -0.07
M/c bond lengths: 98.8% within
limits 1.2% highlighted
* M/c bond angles: 92.5%
within limits 7.5% highlighted
1 off graph
Planar groups:
100.0% within limits 0.0% highlighted
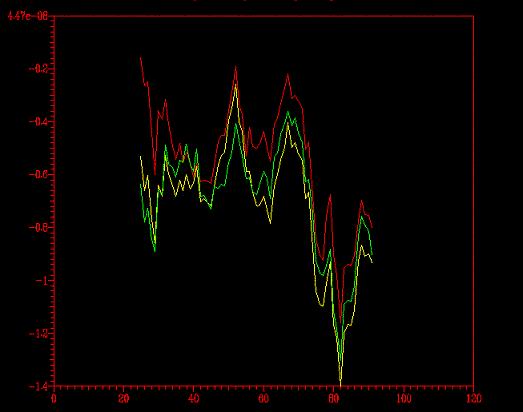
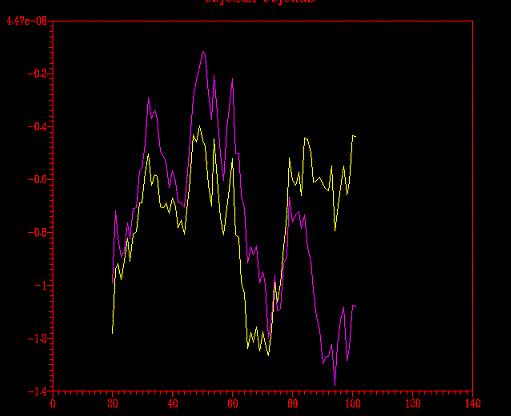
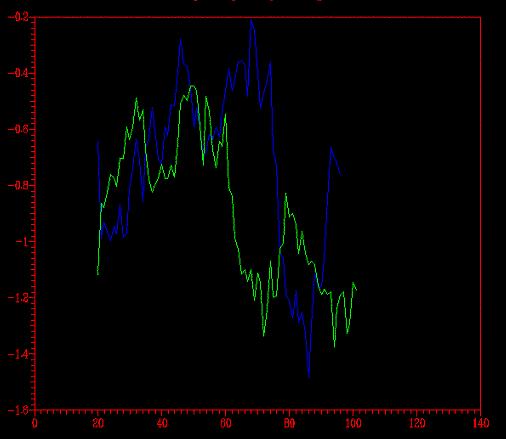
Ahora queda la prueba del perfil (energético, que no psicológico,
aunque también nos indique qué estructura es más estable).
Del programa Prosa extraímos el gráfico siguiente.
Ahora ya sólo nos queda optimizar el modelo y someterlo a una sesión de bailoteo para examinar su dianámica molecular.
La optimización se realizó con el progarma Grumos, repitiendo
el proceso que segumos con el modelo modcut
pero
con los archivos correspondientes, ¿eh? La estructura que resultó
de la superposición fue la más bonita del mundo , y para
que lo veias con vuestros propios ojos, aquí la teneis:

Es tan bonita....
Y para que veais que esto de que es tan bonita no es sólo amor de
madre, realizamos un nuevo Procheck (ya no tiene bad contacts), y los comparamos
con los perfiles energéticos de los otros modelos.
<<< P R O C H E C K S U M M A R Y >>>
floricuaAoptim.pdb 2.8 121 residues
* Ramachandran plot: 77.8% core 20.4% allow 0.9% gener 0.9% disall
All Ramachandrans: 10 labelled residues
(out of 119)
Chi1-chi2 plots:
1 labelled residues (out of 77)
Main-chain params: 6 better
0 inside 0 worse
Side-chain params: 5 better
0 inside 0 worse
Residue properties: Max.deviation:
4.7 Bad
contacts: 0
Bond len/angle: 3.8 Morris et al class:
1 1 2
1 cis-peptides
G-factors
Dihedrals: -0.42 Covalent: 0.11
Overall: -0.20
M/c bond lengths: 99.8% within limits
0.2% highlighted
M/c bond angles: 94.6% within limits
5.4% highlighted
* Planar groups: 60.4% within limits
39.6% highlighted 11 off graph
Se comparó el perfil energético del modelo optimizado con
el del no- optimizado (primera imagen)
y con el del modelo florexA.pdb (segunda
imagen).
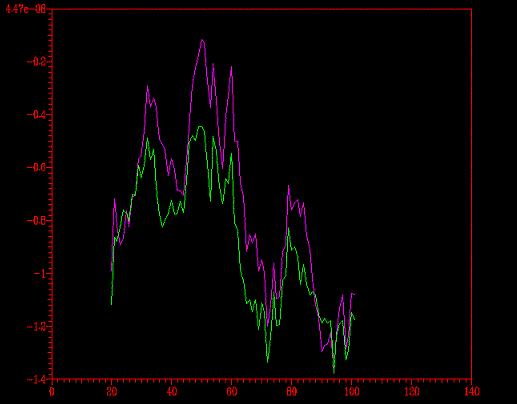



{kind=link}