MODEL
DEFINITIU
CONSTRUCCIÓ DEL MODEL DEFINITIU
El model
estructural definitiu es va construir amb: ht0B, axeA, kevC, ykfD.
![]() Els pdbs de les proteïnes template es van
alinear estructuralment amb STAMP,
l’arxiu obtingut es transformà a format clustalw i després a format sp per
visualitzar-lo al Rasmol .
Els pdbs de les proteïnes template es van
alinear estructuralment amb STAMP,
l’arxiu obtingut es transformà a format clustalw i després a format sp per
visualitzar-lo al Rasmol .
![]() A partir del fitxer obtingut amb el Stamp
modificat s’utilitzà per fer una matriu HMMER
(build, calibrate i align) per trobar un perfil estructural de les ADH, a
partir de les homòlogues finalment escollides, que llençàrem contra la
seqüència ALCEU. Aquest fitxer s’havia de transformar a format clustalw i
després a format pir per poder-lo introduir en el programa modeller .
A partir del fitxer obtingut amb el Stamp
modificat s’utilitzà per fer una matriu HMMER
(build, calibrate i align) per trobar un perfil estructural de les ADH, a
partir de les homòlogues finalment escollides, que llençàrem contra la
seqüència ALCEU. Aquest fitxer s’havia de transformar a format clustalw i
després a format pir per poder-lo introduir en el programa modeller .
![]() El programa MODELLER
llegeix un arxiu top on s’especifica la seqüència problema, els templates en
format pdb i el fitxer en format pir per tal d’obtenir el model estructural de
la proteïna alceu.
El programa MODELLER
llegeix un arxiu top on s’especifica la seqüència problema, els templates en
format pdb i el fitxer en format pir per tal d’obtenir el model estructural de
la proteïna alceu.
![]() Es van generar 15 models dels quals es va
analitzar l’energia amb el PROSA i se’n
van escollir els dos millors (alceu1 i 4), aquests tenien una energia de 0.05 i
0.1.
Es van generar 15 models dels quals es va
analitzar l’energia amb el PROSA i se’n
van escollir els dos millors (alceu1 i 4), aquests tenien una energia de 0.05 i
0.1.
![]() I els millors models també van ser
visualitzats amb RASMOL per saber
l’estructura secundària de la regió del loop definitiva: alceu 1 tenia 2a i 2b,
i alceu 4 tenia 2a i 3b
(1 és d’un residu). (noualceu1est
i noualceu4)
I els millors models també van ser
visualitzats amb RASMOL per saber
l’estructura secundària de la regió del loop definitiva: alceu 1 tenia 2a i 2b,
i alceu 4 tenia 2a i 3b
(1 és d’un residu). (noualceu1est
i noualceu4)
![]() Finalment,
aquests models es van analitzar amb el programa PROCHECK
que edita uns mapes de Ramachandran (format sp) i un fitxer (format sum), on
s’especifica els residus afavorits (core), permesos (allowed), generosament
permesos (gener), no permesos (disallowed) i bad contacts. Els resultats van
ser:
Finalment,
aquests models es van analitzar amb el programa PROCHECK
que edita uns mapes de Ramachandran (format sp) i un fitxer (format sum), on
s’especifica els residus afavorits (core), permesos (allowed), generosament
permesos (gener), no permesos (disallowed) i bad contacts. Els resultats van
ser:
-
ALCEU 1: core (88.9%), allowed
(9.8%), gener (1.8%), disallowed (0.3%) i bad contacts (8). (ALCEUest.sum) (alceuest01.ps)
-
ALCEU 4: core (87.2%), allowed
(9.8%), gener (2.6%), disallowed (0.3%) i bad contacts (8). (ALCEUsum) (alceu01.ps)
Es
va comprovar quants d’aquests residus no permesos pertanyien a la regió del
loop que tenia energia positiva i quina estructura hi havia en aquesta regió.
Aquests residus eren:
-
alceu 1: gln105 (alfa), leu110 (loop), ser113 (no
especificat), gly114 (no especificat), ala125 (beta)
-
alceu 4: leu110 (loop), ser113 (no especificat),
ala125 (no especificat, a l’extrem d’una beta), asn131 (loop)
Els
residus leu110, ser113 i ala125 coincidien en tots dos models perquè deurien
ser els pitjor modelats de la regió del loop.
OPTIMITZACIÓ
L’optimització
es va dur a terme amb el programa GRUMOS.
En
un pas previ es va analitzar si les cisteïnes (cys) dels
models feien ponts disulfur mitjançant el programa VMD.
Les distàncies mesurades entre les cisteïnes eren superiors a 2.5 – 2.8, per
tant, no es pot considerar que es formin ponts disulfur entre les nou cisteïnes
de la seqüència.
Llavors
es va fer l’optimització d’energia pels dos millors models (alceu1 i 4). Les
característiques més importants van ser:
-
sense ponts disulfur, perquè les cisteïnes estaven
massa separades entre elles
-
sense aigua, per facilitar l’anàlisi, encara que el
modelatge amb aigua hagués estat més real perquè la proteïna sempre està en
solució
-
sense periodicitat, perquè no es tractava un
cristall cristal·logràfic
-
sense restriccions de distàncies, de dihedral ni de
posició
El
primer i l’últim arxiu (format gsf) optimitzats de cada model es van superposar
amb el XAM per veure si hi havia
diferències estructurals i obtenir un arxiu pdb amb tots dos.
Degut
a que l’últim arxiu gsf de cada model és el més optimitzat, es va escollir
aquest per analitzar l’energia amb el PROSA
comparant-los amb el model abans d’optimitzar (alceu1 i 4). En el model
optimitzat d’alceu 4 l’energia arribava a zero i en el d’alceu 1 l’energia
tenia valors negatius (alceu1i4opt).
Finalment
es compararen el millor model optimitzat (alceu1opt) amb el mateix model sense
optimitzar (alceu1) i amb el template kev que s’utilitzarà per fer el model
d’alceu amb cofators (sessiontotdefcmd),
veure l’apartat Construcció de models amb cofactors.
Finalment, aquests models es van analitzar amb el programa
PROCHECK
per visualitzar amb el programa GHOSTVIEW
els mapes de Ramachandran (format sp) i un fitxer (format sum), on s’especifica
els residus afavorits (core), permesos (allowed), generosament permesos
(gener), no permesos (disallowed) i bad contacts. Els resultats van ser:
-
ALCEU1 optimitzat: core (78.4%),
allowed (19.3%), gener (1.3%), disallowed (1.0%) i bad contacts (0). (alceuopt1.sum) (alceu1op.ps)
-
ALCEU4 optimitzat: core (75.4%),
allowed (21.3%), gener (1.6%), disallowed (1.6%) i bad contacts (0). (alceu4op.sum) (alceu4op.ps)
Amb
l’optimització, encara i disminuir els residus favorables i augmentar els
desfavorables, van disminuir els bad
contacts a zero, que era el que es volia aconseguir, ja que els residus en
posició desfavorable s’espera que disminueixin amb la dinàmica.
L’alceu
1 optimitzat tenia energia menor i millors tants per cents en els residus
disallowed i en el core, que alceu 4 optimitzat. Per tant, es va escollir l’ alceu1 com a model definitiu.
DINÀMICA
Es
va fer la dinàmica del model alceu 1 optimitzat amb el programa GRUMOS.
Les
característiques més importants van ser:
-
amb SHAKE per constrenyir
distàncies
-
sense restricció de posició, distància o dihedral
Seguidament
es realitzà l’anàlisi dels 10 fitxers (format hs) i els 10 fitxer (format gsf)
es van superposar amb el XAM per obtenir
un fitxer pdb amb tots els models de la dinàmica.
Mitjançant
el PROSA es va analitzar l’energia
d’aquestes noves conformacions. El patró d’energia mostrava una fluctuació
d’energia en la regió del loop sent major de cero en algunes conformacions i
menor de cero en altres (sessionmd.cmd). No es
pot dir que en la regió del loop, la conformació adoptada amb menys energia
sigui més estable perquè no se sap en quines condicions és més estable la proteïna
alceu. En les altres regions la proteïna està molt conservada.
B-FACTOR
L’opció
B-factor dins del programa GRUMOS permet
veure la fluctuació de l’estructura segons la temperatura d’anàlisi. La
temperatura està directament relacionada amb la velocitat de moviment dels
àtoms, les regions amb alta temperatura són aquelles que fluctuen més.
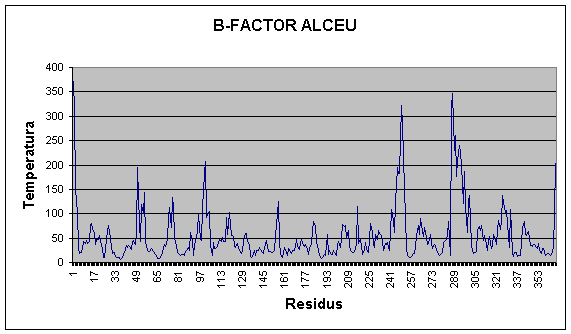
Es
van analitzar els resultats obtinguts en el B-Factor: (bfactorpics)
·
El pic de 289-300 corresponia a un tros del
plegament Rossman del model alceu. Les estructures trobades en aquests
aminoàcids eren un tros de beta trencada, un tros de loop i un d’hèlix alfa. En
aquesta regió hauria d’haver-hi una beta del plegament Rossman implicada en el
domini de dimerització, per tant, el modelatge no és del tot correcte.
·
El pic de 243-255 també pertanyien al mateix
tipus de plegament on s’observava una hèlix alfa trencada.
·
El pic de 100-110 pertanyia a una cadena
beta mal modelada que no se sap a quina estructura ha de correspondre realment
ja que ha estat impossible modelar la regió del residu 90-130 (analisiloop)
·
El pic de 49-57 corresponia a un tros de
loop, una beta d’un residu i un tros no especificat. Se suposa que aquesta
regió hauria de correspondre a una regió de loop.
