OBJECTIUS
Fes el modelat de la subtilisina i un anàlisi comparatiu d’estructures de la família amb la que has fet el model.
Pràctica 5.1
Per tal de construir un model, necessitem trobar seqüències homòlogues en les que ens basarem per a la seva elaboració. En aquesta primera pràctica realitzarem la búsqueda d'aquestes seqüències, i escollirem aquelles que considerem adients, per a posteriori construir el model.
Tenim la seqüència de la proteïna subtilisina P11018.fasta (proteïna problema). Farem una búsqueda de seqüències homòlogues mitjançant el PSI-BLAST. Ens interessa obtenir els homòlegs de PDB, ja que necessitem l'estructura de les proteïnes cristalitzades per a realitzar el model. Primer de tot utilitzarem PSI-BLAST contra swissprot, i la matriu que obtindrem la utilitzarem per a fer la busqueda d'homolegs a PDB mitjançant un altre cop PSI-BLAST. Ho fem així perquèla base de dades de swissprot es més gran que pdb, i així obtenim una matriu de busqueda molt mes acurada. Fent-la servir contra PDB, obtindrem seqüències que no obtindríem si féssim el PSI-BLAST directament contra PDB. PSI-BLAST, al fer la cerca mitjançant matrius de pesos, obtindrà uns resultats més sensibles que BLAST. A més, ens permetrà fer la búsqueda amb Swissprot i posteriorment utilitzar la matriu obtinguda, per cercar en PDB.
Correm el PSI-BLAST contra swissprot, amb dos iteracions. Obtenim la matriu blosum p11018.bls1 .
[e...]$ blast -i p11018.fasta -d /disc9/DB/blast/swissprot -C p11018.bls1 -j 2
Correm el PSI-BLAST contra PDB.
[e...]$ blast -i p11018.fasta -d /disc9/DB/blast/pdb -R p11018.bls1 -C p11018.bls2 -o p11018.psiblast -j 2
Obtenim com a outputs de PSI-BLAST:
Dins el fitxer p11018.psiblast tindrem la llista de seqüències ordenades per homologia. Escollirem les seqüències adients per a construir el model. Les seqüències que he escollit no són les de més homologia. Si escollís seqüències de molta homologia, el model estaria realitzant el model amb la mateixa proteïna problema. He escollit les següents proteïnes segons el seu E value:
Pràctica 5.2
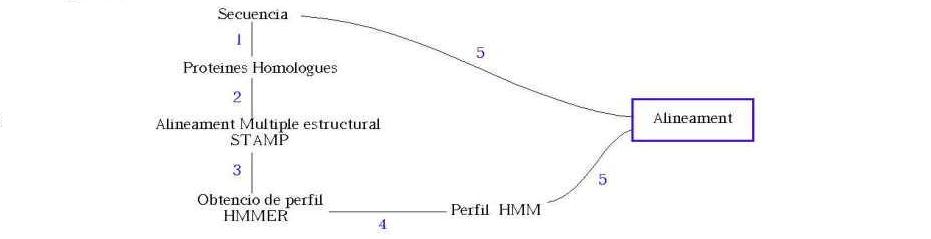
Alinearem les estructures, extraurem un perfil que tindrà tota la informació estructural. Les estructures son més conservades, i la informació que extraurem serà més valuosa que amb els alineaments de seqüències.
A partir de les seqüències utilitzades en l'alineament de la practica 5.1, obtindrem els fitxers amb les estructures (pdb), i realitzarem l'alineament de les estructures, amb el programa STAMP.
Pràctica 5.3
En aquesta pràctica construirem definitivament el model mitjançant el programa MODELLER.
Previ a l'execució del programa, necessitem l'alineament de les proteïnes.
Utilitzarem l'alineament d'estructures amb HMMER (hmm.ali, de la practica5.2), i l'alineament de seqüències fet amb Clustalw a partir de les seqüències trobades amb PSI-BLAST (llista.aln, de la practica5.1, però que per a no crear confusions he anomenat llista.ali)
Aquests alineaments els hem de transformar en un tipus d'alineament que entengui el MODELLER. Utilitzarem l'script aconvertMod2:
$ aconvertMod2.pl -in h -out p < hmm.ali >hmm.aln
Per tant, per a crear el model utilitzarem els alineaments hmm.aln i llista.aln.
De l'alineament haurem d'esborrar tots els caràcters estranys (exemple: a la part de la seqüència P11018, haurem d'esborrar tots els signes de la capçalera, innecessaris)
Per executar el modeller necessitem:
Per executar el modeller, no he pogut utilitzar la seqüència 1a2q , ja que produia errors. He intentat tornar a baixar el seu pdb, però continuava donant errors. Finalment hem obtingut 4 models, dos per l'alineament Clutsalw i altres dos per l'alineament HMM:
model1.hmm
model2.hmm
model1.clu
model2.clu
Visualització :podem veure els models obtinguts mitjançant Rasmol.
Observem al visualitzar el models, que de cert mode, sobra cadena. Si observem l'alineament de les seqüències, llista.ali, veiem que es normal que sobri, ja que la seqüència problema és més gran que les templates. Haurem de tallar el PDB, en els extrems per a que el model sigui correcte. Els fragments tallats son els següents:
Després d'haver analitzat els models, observant-los amb ProsaII (Practica 6), m'he adonat que en dos regions la energia era positiva. Mirant l'alineament de Clustalw, he deduït que aquests pics podrien ser deguts als gaps que introdueix la seqüència 1svn (gaps). Per tant he tornat a realitzar el model (i tota la pràctica 5), treient del model la seqüència 1svn amb l' intenció de millorar-ho.
Els nous arxius obtinguts, al realitzar el nou model són:
Alineaments:
HMM
ClustalW: observem que els gaps abans problemàtics han desaparegut. Però encara queden altres que no es poden treure, o que si aconseguim treure'ls provocaríem l'aparició d'altres gaps.
Models:
model1.hmm
model2.hmm
model1.clu
model2.clu
{kind=link}