OBJECTIUS
Validació i identificació de regions mal modelades del model de subtilisina de la prąctica 5. Arreglar l’aliniament amb la predicció d’estructura secundąria i mirar de millorar els models.
Prąctica 6.1
PROCHECK
Analitzarem el model mitjanēant PROCHECK. L'objetiu de Procheck és valorar la 'calitat estereoquķmica' de l'estructura de la proteļna donada, comparant amb estructures ben refinades a la mateixa resolució, i dona una indicació de la fiabilitat de cada residu. Per a fer aquesta valoració, el programa utilitza diferents parąmetres. Sobretot, el parąmetre que nosaltres mirarem serà el plot de Ramachandran mirant els angles phi-psi.
Hem de mirar la resolució mes alta (per tant, pitjor resolució) dels pdb's utilitzats al fer el model.
execució:
$ procheck_single model1.hmm 2.0
Obtenim els arxius .ps
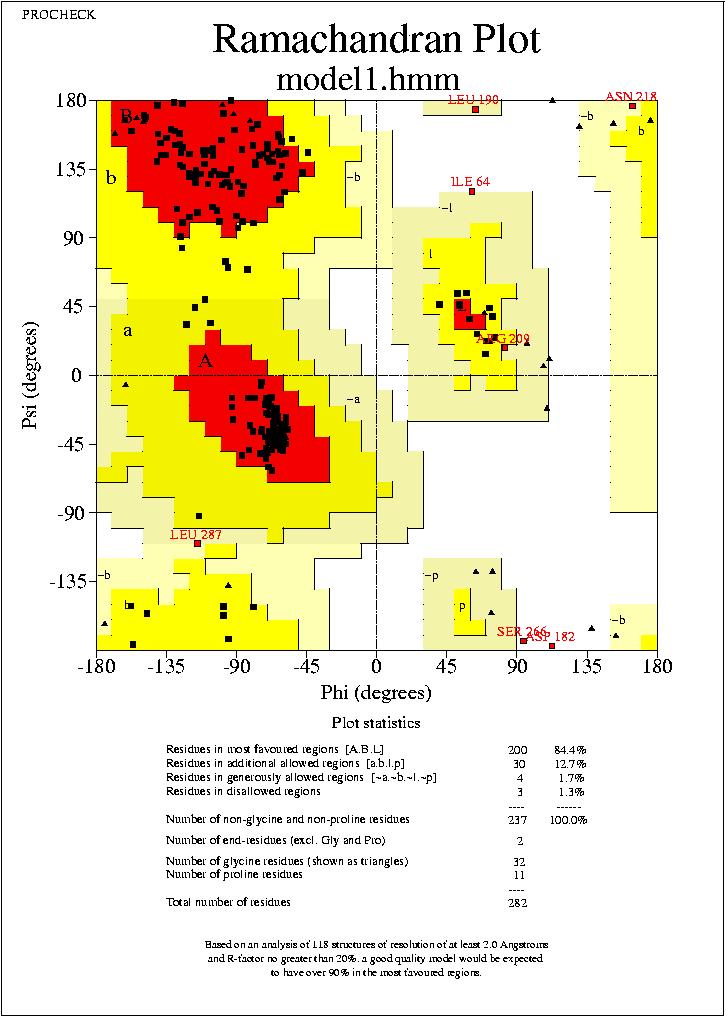
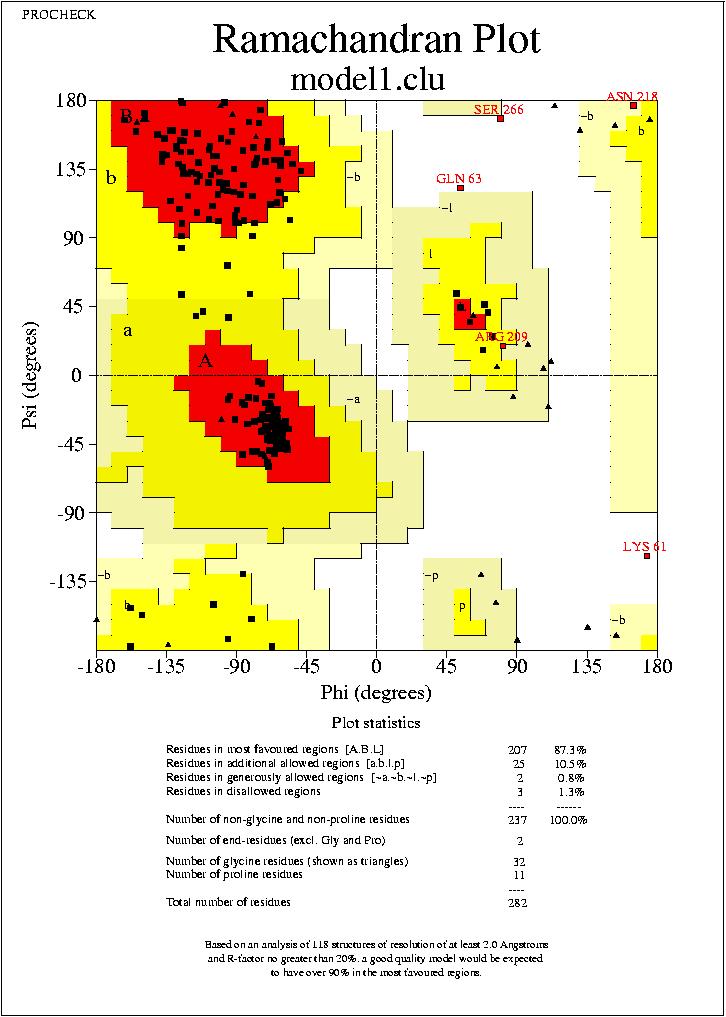
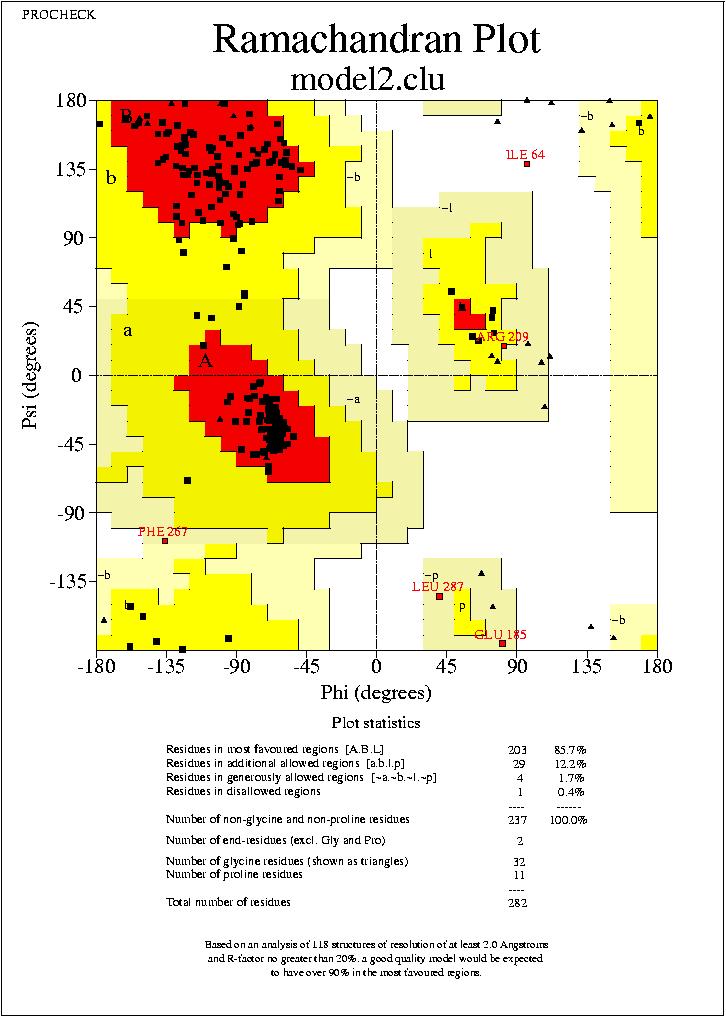
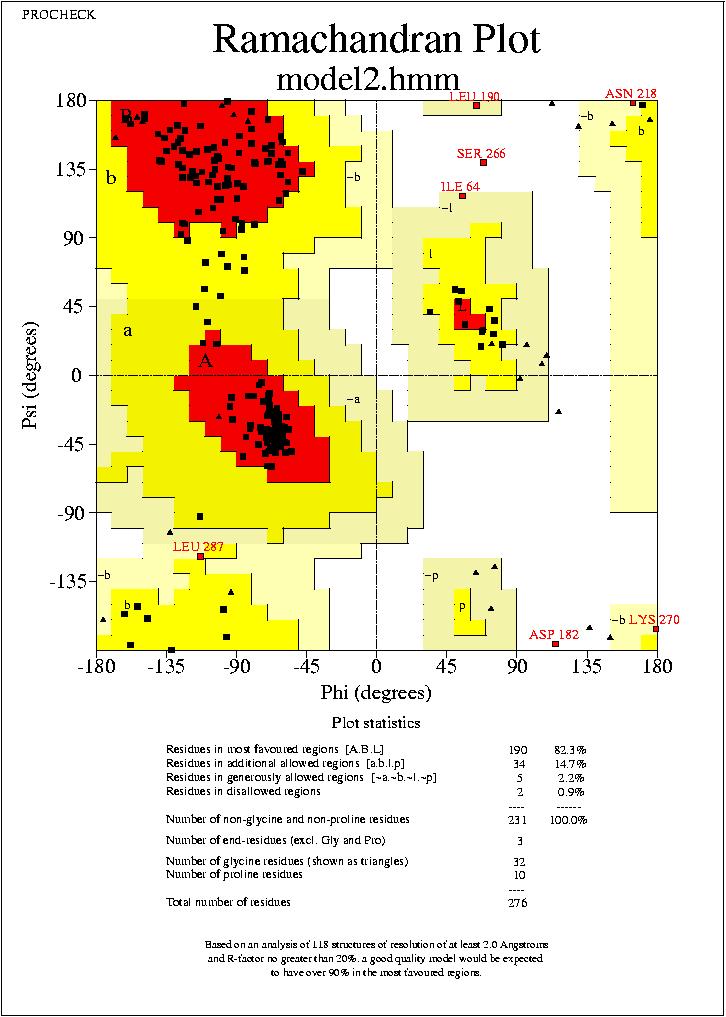
Podrem visualitzar aquests arxius mitjanēant el programa Ghostview. Podrem visualitzar els mapes de Ramachandran de tots quatre models, visualitzant els arxius:
model1.hmm_01.ps,
model1.clu_01.ps,
model2.clu_01.ps,
model2.hmm_01.ps.
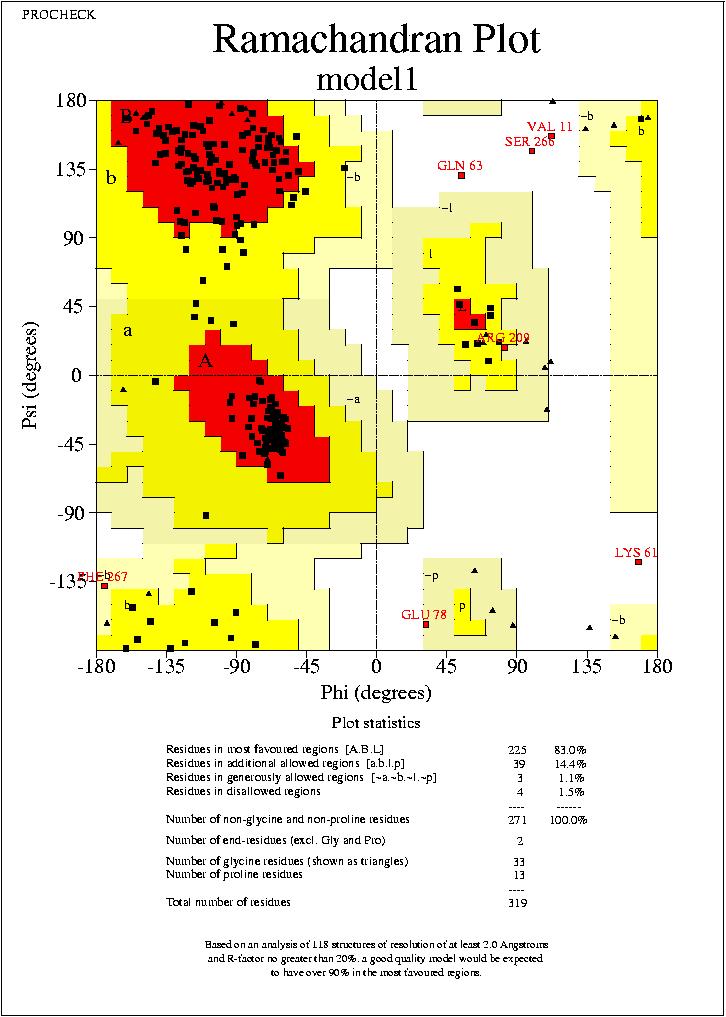
No puc extreure conclusions clares dels resultats observats al mapa de Ramachandran, ja que els mapes dels quatre models son molt similars, amb petites variacions, i en tots els casos el percentatge de residus dins de les regions favorables (Residues in most favoured regions) i dins de les regions permeses (Residues in additional allowed regions)
,supera el 90%. Només podem veure, que els segons models (tant de HMM com de Clustalw) tenen un resultat més favorable.
Haurem d'esperar a fer l'anąlisis amb Prosa, per concluir quin es el millor model.
DSSP
El programa DSSP defineix l'estructura secondąria, les caracterķstiques geomčtriques i l'exposició de les proteļnes al solvent, donant les coordenades atomiques en format PDB. El programa no fa una predicció de l'estructura secondąria.
El fitxer d'Input haurà de ser el pdb amb les coordenades atņmiques. Obtenim el fitxer model2.hmm.cut.dssp.
$ dssp P11018.B99990001 model.hmm.cut.dssp
ALISS
L'estructura obtingua de DSSP, la podem transformar. ALISS (Alingment Independent Similarity Searcher) detecta eficaēment similituts locals entre dos proteļnes. El fitxer obtingut tindrà un format més similar a Clustalw
Prąctica 6.2
ProsaII
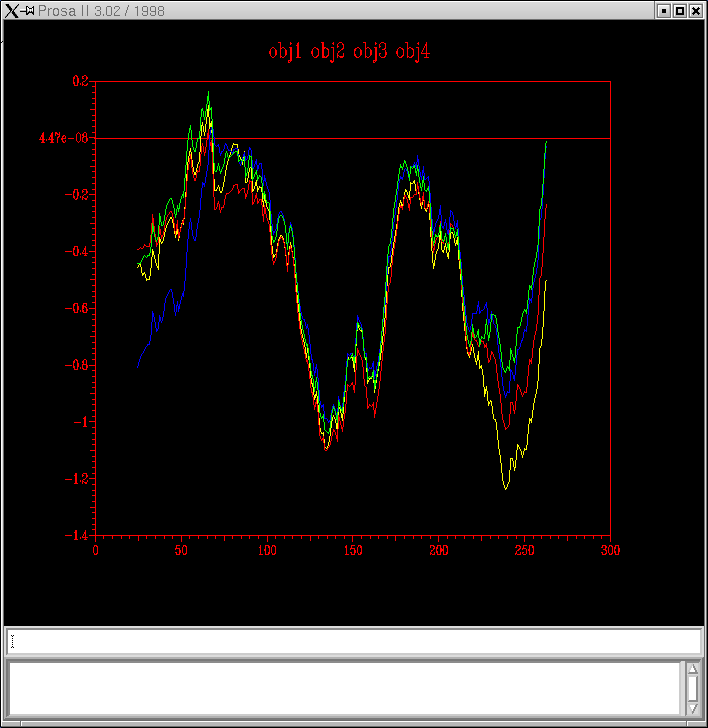
Analitzant la grąfica, podem observar que el model obtingut no serķa massa correcte. Apareixen dos pics de energia positiva, que podrķen correspondre a les zones de gaps observats al alineament inicial de Clustalw.
Anąlisis del nou model:
Procheck:
Prosa:
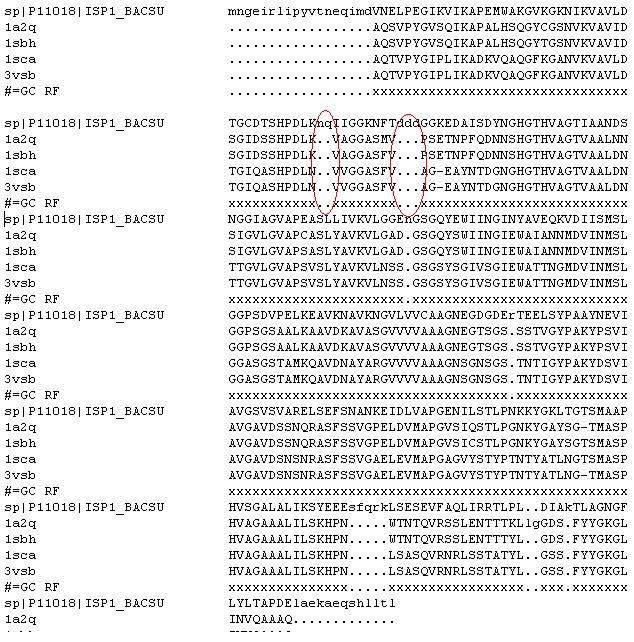
Al analitzar Prosa, sembla ser que inclśs després de tallar, el millor model es el blau (model2.hmm.cut). Però encara te una zona en N-term en la que s'observa un petit pic en el que la E es positiva. Al observar l'alineament sembla ser que pot coincidir amb una zona en la que en l'alineament (hmm.ali) hi ha gaps. Fem un Stamp de una de les estructures templates amb el model model2.hmm.cut.
Prąctica 6.3
GROMOS
Avaluació del model
Executem Grumos
Mirem:
XAMinicial i final(factor multiplicació 10):
PROCHECK
Prosa ens ajudara a analitzar si el nostre model és correcte. Prosa calcula un score (Z-score) del nostre model. El score indicarà la calitat de l'estructura. Els scores dels plegaments de les estructures natives tenen uns rangs caracterķstics. Si l'score obtingut està fora del rang, llavors l'estructura pot tenir problemes.
Amb Prosa podem obtenir grąfiques d'energia del model, que ens ajudarà en l'anąlisis de les diferents regions. Aquests grąfics mostren la arquitectura energčtica del plegament de la proteļna en funció de la posició dels aminoącids en la seqüčncia. Utilitza la inversa de la llei de Boltzmann per obtenir energies. Si observem altes energies a la grąfica, podem deduir que es troba d'una secció massa 'forēada' que pot indicar una part problemątica en el plegament. D'aquesta forma podem millorar el model.
Prosa analitza el model mitjanēant els potencial estadķstics. A partir de les distancies entre cada aminoącid,
conta la probabilitat de que dos aminoącids es trobin a una distancia concreta en l'espai.
execució:
Per tal d'executar el prosa, i veure un grąfic amb els 4 models, he realitzat un script de l'execució:prosa.ss
$ prosa -s prosa.ss
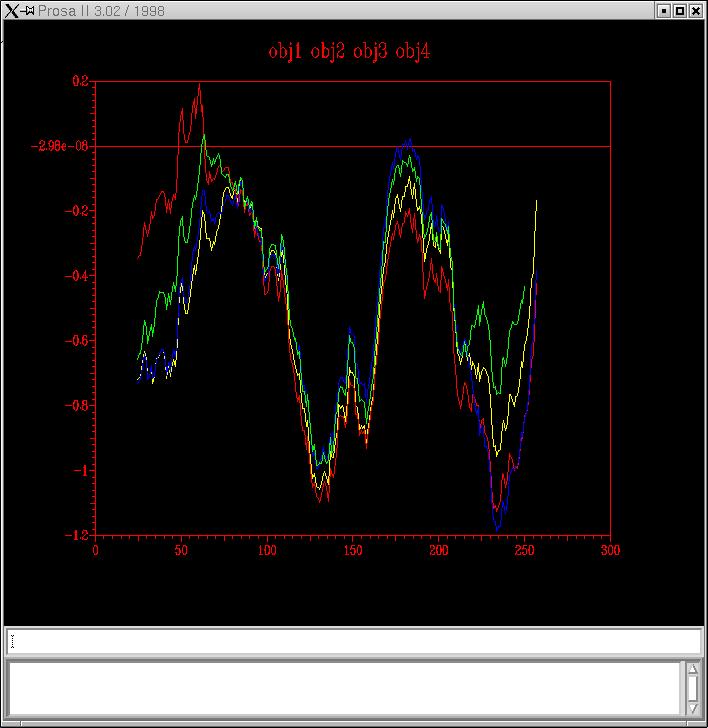
Obtenim aquest grąfic:prosa
A més, la lķnia que correspondria al model2.hmm (lķnia vermella), és la que en una de les zones sobrepassa més ampliament el 0. En teoria, aquest model hauria de ser el més correcte, però no és aixķ. Una posible explicació podria ser que al haber tanta homoloiķa entre les seqüčncies, l'alineament entre seqüčncies serķa més correcte que l'alineament estructural. Però en el meu cas, no hauria de ser aixķ, per que tal i com he explicat a la practica 5, les proteļnes que he escollit per a realitzar els models no són de les que major homologia presentaven.
Finalment, he decidit tornar a fer el model. Mirant l'alineament de Clustalw, observem dos zones de (gaps) que podrķen ser eliminades si treķessim la seqüčncia 1svn. Tornare a realitzar la Practica 5 , però sense utilitzar aquesta proteļna com a template.
model1.hmm_01.ps
model1.clu_01.ps
model2.clu_01.ps
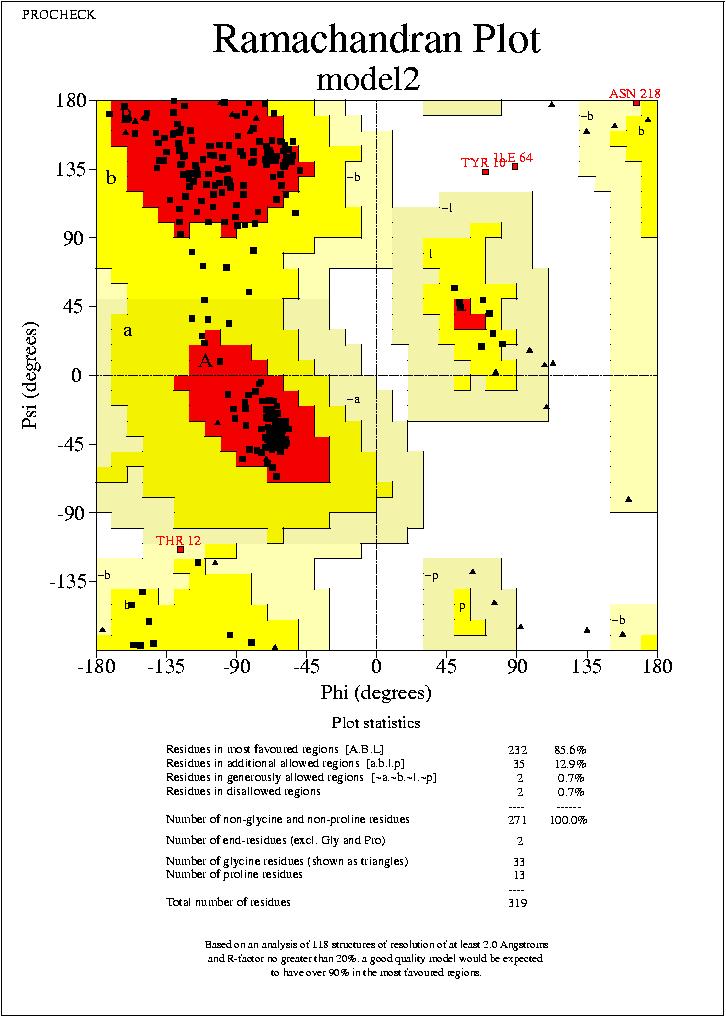
model2.hmm_01.ps
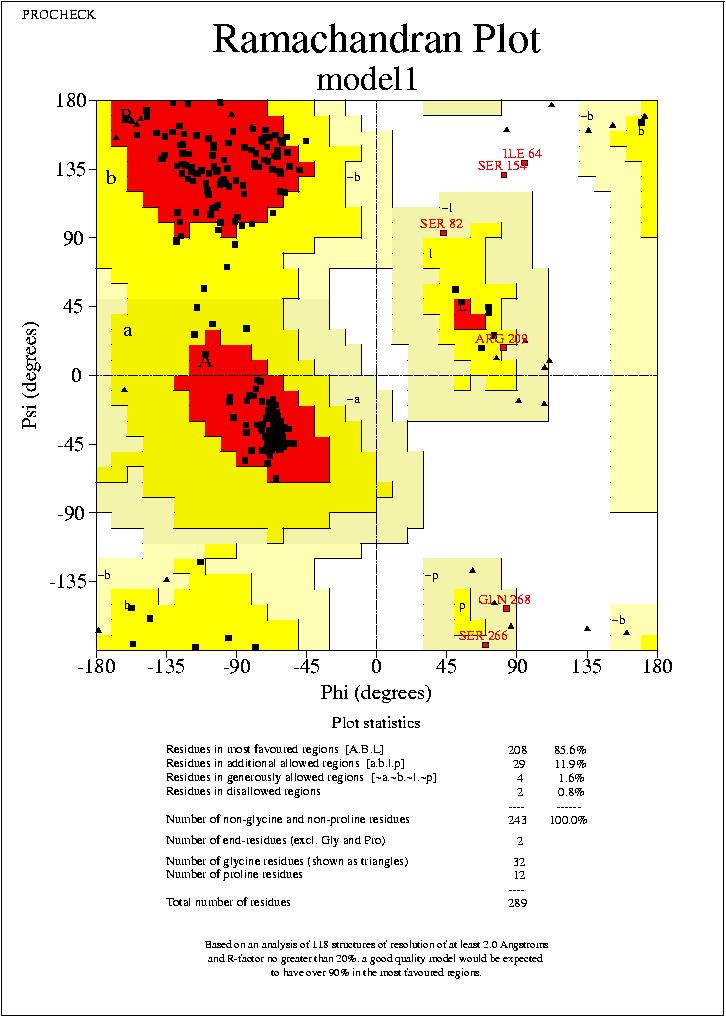
Els resultats del plot de Ramachandran es similar al del anterior model. En alguns casos podrķem dir que el nou model seria pitjor. Però encara no podem arribar a cap conclusió, sense realitzar el Prosa.
Grafic de Prosa del nou model.
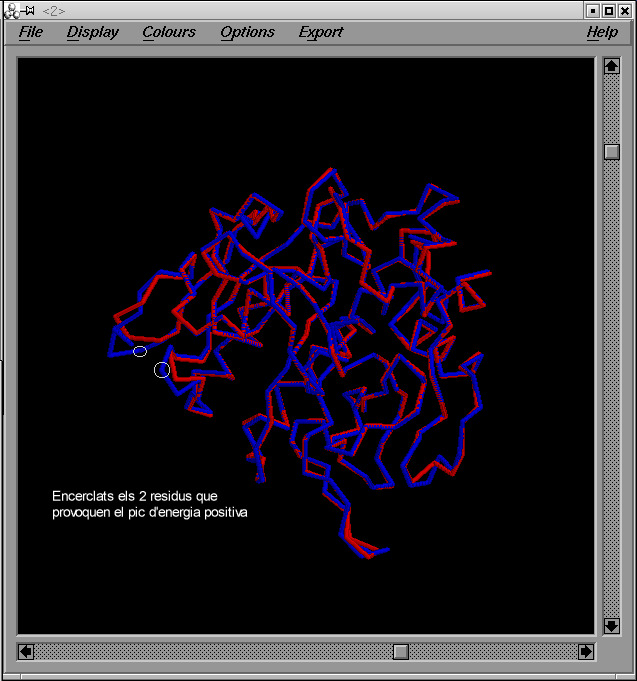
Podem veure la imatge de la superposició : prueba1pdb. En aquest imatge estan assenyalats els ątoms dels que hem fet referčncia.
Amb aquesta superposició podrem observar les diferčncies entre la template i el model. Observem dos zones de loops, que són més llargues en el model. Això produeix que els loops estiguin més a prop en el model. Si mirem els atoms dels loops del model, que estan més a prop veiem que son: GLU 122 - ASP 72, els dos tenen cąrrega negativa. Si tenen igual cąrrega i estan tant a prop, la energia que observem a Prosa, serà positiva. La distąncia a la que es troben és: 14'470 A.
Possiblement el pic d'energia positiva que s'observa, vingui donada per aquest fet.
GROMOS és un programa que serveix per a realitzar el que anomenem optimització geomčtrica. Per a optimitzar un model cal buscar els mķnims energčtics (valls). Hi ha varis factors a tenir en compte: la distąncia d'interacció, l'angle d'enllaē, el diedre,(components enllaēants), i les forces de Van der Waals i interaccions electroestątiques, (components no enllaēants). Si tenim en compte tot aixņ, obtenim l'equació del camp de forces, que ho ajunta tot i permet que si l'optimitzem millori l'energia del model.
Correccions: Al generar el model creem torsions, tensions i altres desajusts per tal de que la seqüčncia problema s'ajusti als templates. Aquests forēaments són el que prosa, procheck, etc... ens mostra i és precisament això el que cal corregir.
Per a familiaritzar-nos amb GRUMOS, hem fet l'exemple: pci.pdb.
En el model ja havķem afegit la terminació TER i OXT, necessaris per a l'execució de GRUMOS. Fem els passos previs per a l'execució de GRUMOS:
Canviem el nom del model model2.hmm.cut a model2.hmm.pdb
$ cp model2.hmm.cut model2.hmm.pdb
Executem arrangeG
$arrangeG.pl modelo.pdb modeloG.pdb
$arrangeG.pl model2.hmm.pdb model2G.hmm.pdb
Afegim el TER ( ja que l'arrangeG, ens el treu)
Mirem amb Rasmol si existeixen ponts disulfurs:
select cys
color CPK
pick distance
Les dos cļsteines trobades es troben a més de 4A, i per tant no podran formar ponts disulfur
Atom #1: CYS35.CA (256)
Atom #2: CYS157.CA (1137)
Distance CYS35.CA-CYS157.CA: 17.505
energia inicial: 0.12119E+10
energia final: -0.15359E+05 : model2G.hmmxemnum010.gsf
Utilitzem el xam per fer la superposició d'estructura del model que tenķem abans d'executar Grumos(model2G.hmm.pdb) i el que ens ha sortit de la optimització (model2G.hmmxemnum010.gsf )
D'aquesta forma aconseguim veure la superposició ( i per tant, veure les diferencies) i convertir model2G.hmmxemnum010.gsf a pdb
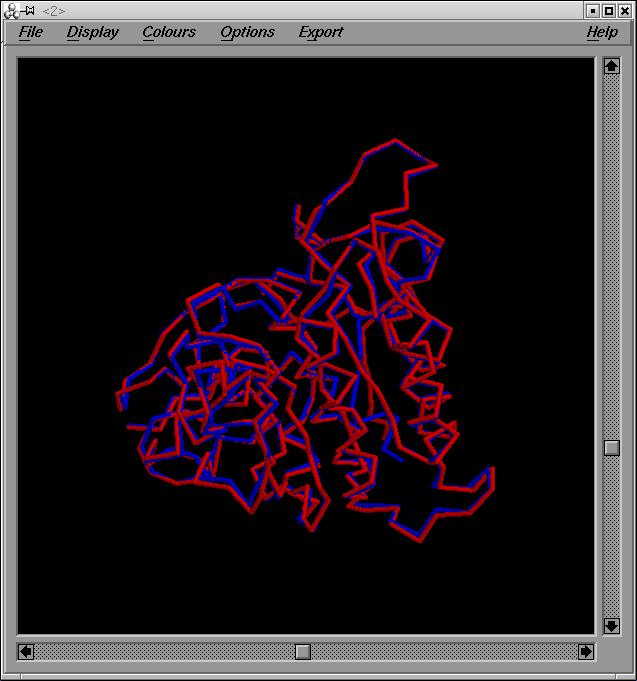
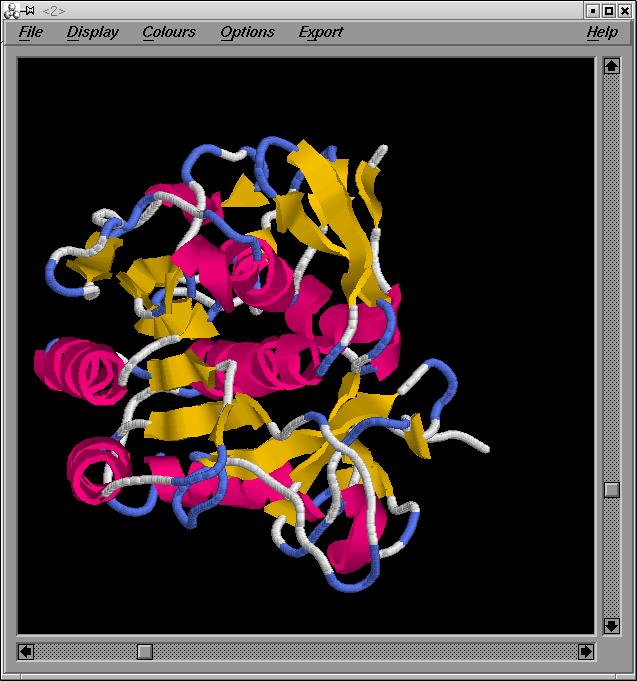
El pdb q obtenim de la superposició [modelfin.pdb ], hem d'esborrar la primera part (que correspondrà al pdb q teniem abans de grumos- model2G.hmm.pdb). Podem veure una imatge de la superposició: superposició . El nou pdb el guardo com a model.final.pdb.
Podem veure una imatge del model final: model.final
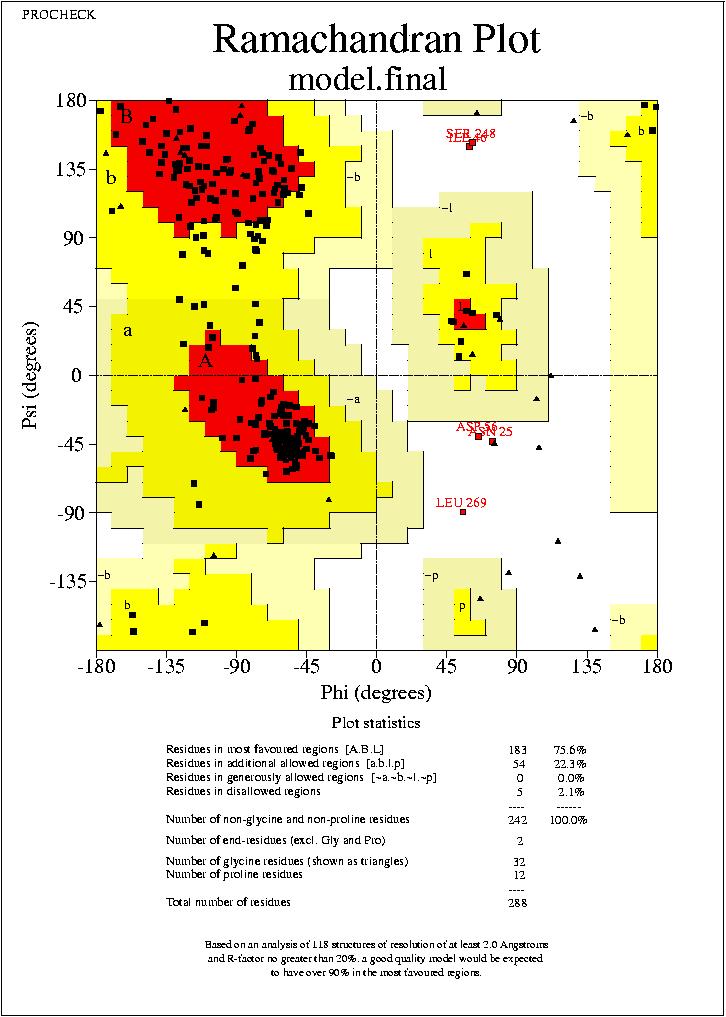
rms superposició (rms del arxiu modelfina). El valor del Procheck, no sembla massa correcte, i ens porta a dubtar de la correcció de la superposició.
Realitzem un procheck del nou model, per comparar amb el model sense optimitzar:
model.final_01.ps
model2.hmm_01.ps
Podem veure, com clarament, els valors que s’ obtenen després de l’ optimizació, son pitjors que els del model previ. Això pot ser degut a que en la optimizació s’han produļt canvis en aminoącids que produeixen aquests resultats.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}