|
|
- DESCRIPCIÓ TEÒRICA DEL PROGRAMA
En la primera part de la pràctica, utilitzarem el programa XAM, per a realitzar aliniaments estructurals entre les diferents proteïnes analitzades.
XAM és un programa dÇaliniament estructural que realitza una superposició de dues proteïnes tot realitzant tant moviments de rotació (per a encaixar les estructures) com moviments de translació (mou les estructures fins a superposar-les).
Aquest programa tracta de comparar cada un dels residus dÇuna estructura amb els residus de lÇaltra (per exemple, agafa el residu 1 de la cadena A i el compara amb el primer residu de la cadena B, igual amb el residu 2...), per tal dÇintentar trobar identitat entre ambdues. LÇordre dels diferents àtoms que pren per a cada estructura, és lÇordre existent en el seu corresponent arxiu .pdb.
De la comparació dels diferents residus seÇn calcula un valor de RMSD (Root Mean Square Deviation function), que ens ve a indicar com de semblants són dues estructures.

- DESCRIPCIÓ PRÀCTICA DELS COMANDAMENTS DEL PROGRAMA I EXEMPLES DE COMANDAMENTS I CONDICIONS APLICADES EN LÇEXECUCIÓ
Les comandes utilitzades durant aquesta part de la pràctica, han estat:
$ cp /disc9/practica_4/XAM .
$ cd XAM
$ ls
helix1.pdb
helix2.pdb
helix3.pdb
helix4.pdb ...
Un cop executades aquestes ordres, es tracta dÇanalitzar lÇestructura, el nombre de residus i demés característiques de les quatre hèlixs donades com a exemple, per a finalment arribar a lÇexecució del programa de superposició dÇestructures en qüestió (xam).
Una comanda interessant per a conèixer el nombre de residus dÇuna determinada proteïna, és:
$ grep CA helix4.pdb > comandanumresidus
I lÇarxiu de sortida (comandanumresidus), mostrarà només els carbonis α de lÇarxiu helix4.pdb:
ATOM 2 CA ARG 1 -10.776 208.517 81.308
ATOM 13 CA ALA 2 -10.322 209.753 77.707
ATOM 18 CA ILE 3 -6.946 208.110 77.497
ATOM 26 CA LEU 4 -8.694 205.023 78.799
ATOM 34 CA VAL 5 -11.632 204.919 76.373
ATOM 41 CA ASP 6 -9.258 205.640 73.481
ATOM 49 CA TRP 7 -7.138 202.576 74.472
ATOM 63 CA LEU 8 -10.328 200.516 74.574
ATOM 71 CA VAL 9 -11.030 201.504 70.957
ATOM 78 CA GLU 10 -7.584 200.169 70.066
ATOM 87 CA VAL 11 -8.041 196.955 72.054
ATOM 94 CA GLY 12 -11.304 196.522 70.174
ATOM 98 CA GLU 13 -9.627 196.947 66.770
ATOM 107 CA GLU 14 -6.766 194.607 67.704
|
Ara passarem a executar el programa i estudiar-ne les seves possibles aplicacions. Com a exemple, realitzarem la superposició estructural entre lÇhelix3.pdb i lÇhelix4.pdb (tenen igual nombre de residus).
[e14910.bio.acexs.au.upf@au48239 practica_4]$ /disc9/Superposition/xam/xam
Output file name : output_3_4
Input file list? or (cr) :
Structure 1 or (cr) : helix3.pdb
Structure 2 or (cr) : helix4.pdb
Structure 3 or (cr) :
Open file:
helix3.pdb
ATOM 1 N LYS 1 -30.968 220.204 69.410
Total residues: 14 total atoms: 115 in structure: 1
Open file:
helix4.pdb
ATOM 1 N ARG 1 -9.673 209.345 81.811
Total residues: 14 total atoms: 115 in structure: 2
Total molecules: 2 total groups: 1
| All mole. are the same type |
, if OK, (cr):
1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML,
7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM,
9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 1
Backbone atoms: N,CA,C,P,O5',C5',C4',C3',O3'
BB atoms are listed above, if yes (cr) :
Cyclopeptide? default is not, if OK (cr) :
RMSD of 1st str. to the rest? default is not:
Output file for superimposed struc.? or (cr):
Output file for mean structure? or (cr) :
Fragments for superposition, (default: all)
first & last residue of frag. 1:
Selected residues for SC? filename or (cr):
[...]
1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML,
7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM,
9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 1
Backbone atoms: N,CA,C,P,O5',C5',C4',C3',O3'
BB atoms are listed above, if yes (cr) :
Cyclopeptide? default is not, if OK (cr) :
RMSD of 1st str. to the rest? default is not:
Output file for superimposed struc.? or (cr): helix_3_4.pdb
Output fmt 1=DG,2=PDB,3=AMBER,4=bPDB,5=GSF,(cr)=DG 4
Key words (4 lettes, e.g., 5PTI) :
A (all atoms) or S (superimposed)? (cr)=A
Output for BB or Heavy superim.? B/H (cr)=B
Output file for mean structure? or (cr) :
Fragments for superposition, (default: all)
first & last residue of frag. 1:
Selected residues for SC? filename or (cr):
[...]
1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML,
7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM,
9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 0
STOP Oh, XAM! statement executed
|
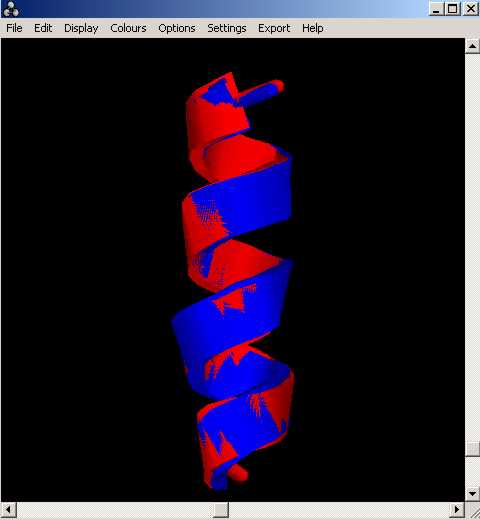
Un cop executat el programa, resulten dos arxius de sortida: un arxiu .pdb (mostra les dues estructures superposades visualment) i lÇaltre, el valor de RMSD (indica com de bo és aquest aliniament). Tot clicant els annexes següents, comprovaràs els resultats obtinguts amb lÇactual exemple: helix_3_4.pdb i output_3_4
Cal tenir en compte la longitud dels residus a superposar, ja que si aquesta llargada difereix entre les diferents estructures a comparar, llavors el programa dóna errors. Més endavant, ja veurem alguns exemples...
- RESULTATS OBTINGUTS DE LÇAPLICACIÓ DEL PROGRAMA ALS EXEMPLES COMENTATS I PROBLEMES PROPOSATS A LA PRÀCTICA
- Calcula el millor RMSD entre les estructures de les hèlixs 1,2,3 i 4. Quins són els intèrvals de residus més adequats per tal dÇobtenir el millor aliniament estructural?.
En aquest cas, executarem de nou el programa XAM, però ara caldrà tenir especial cura del nombre de residus de les diferents estructures a aliniar:
helix1.pdb= 23 residus
helix2.pdb= 18 residus
helix3.pdb= 14 residus
helix4.pdb= 14 residus
Val a dir que els resultats que es mostren a continuació són els millors obtinguts dÇentre les diferents proves realitzades, per tal dÇobtenir el millor RMSD tot agafant el major nombre de residus compartits entre les diferents estructures.
... de nou lÇexecució del programa, mostra:
[e14910.bio.acexs.au.upf@au48239 practica_4]$ /disc9/Superposition/xam/xam
Output file name : totesquatre
Input file list? or (cr) :
Structure 1 or (cr) : helix1.pdb
Structure 2 or (cr) : helix2.pdb
Structure 3 or (cr) : helix3.pdb
Structure 4 or (cr) : helix4.pdb
Structure 5 or (cr) :
Open file:
helix1.pdb
REMARK TITLE
Total residues: 23 total atoms: 199 in structure: 1
Open file:
helix2.pdb
ATOM 304 N SER 37 -9.461 213.395 81.694
Total residues: 18 total atoms: 150 in structure: 2
Open file:
helix3.pdb
ATOM 1 N LYS 1 -30.968 220.204 69.410
Total residues: 14 total atoms: 115 in structure: 3
Open file:
helix4.pdb
ATOM 1 N ARG 1 -9.673 209.345 81.811
Total residues: 14 total atoms: 115 in structure: 4
Total molecules: 4 total groups: 1
1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML,
7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM,
9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 1
Backbone atoms: N,CA,C,P,O5',C5',C4',C3',O3'
BB atoms are listed above, if yes (cr) :
Cyclopeptide? default is not, if OK (cr) :
RMSD of 1st str. to the rest? default is not:
Output file for superimposed struc.? or (cr):
Output file for mean structure? or (cr) :
Fragments for superposition, (default: all)
first & last residue of frag. 1: 7 17
first & last residue of frag. 2:
Selected residues for SC? filename or (cr):
Molecule 2 has different length!
If the fragments for superimp. are different, give the ranges, otherwise (cr)
first & last residue of frag. 1: 39 49
first & last residue of frag. 2:
Molecule 3 has different length!
If the fragments for superimp. are different, give the ranges, otherwise (cr)
first & last residue of frag. 1: 3 13
first & last residue of frag. 2:
Molecule 4 has different length!
If the fragments for superimp. are different, give the ranges, otherwise (cr)
first & last residue of frag. 1: 3 13
first & last residue of frag. 2:
Atom order not checked, because of different molecular
lengthes or identifying
atoms to be displayed!
Atom order not checked, because of different molecular
lengthes or identifying
atoms to be displayed! |
Warning: no calculations for heavy atoms, its number not equal in molecule 2
1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML,
7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM,
9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 1
Backbone atoms: N,CA,C,P,O5',C5',C4',C3',O3'
BB atoms are listed above, if yes (cr) :
Cyclopeptide? default is not, if OK (cr) :
RMSD of 1st str. to the rest? default is not:
Output file for superimposed struc.? or (cr): totesquatre.pdb
Output fmt 1=DG,2=PDB,3=AMBER,4=bPDB,5=GSF,=DG 4
Key words (4 lettes, e.g., 5PTI) :
A (all atoms) or S (superimposed)? (cr)=A
Output for BB or Heavy superim.? B/H (cr)=B
Output file for mean structure? or (cr) :
Fragments for superposition, (default: all)
first & last residue of frag. 1: 7 17
first & last residue of frag. 2:
Selected residues for SC? filename or (cr):
Molecule 2 has different length!
If the fragments for superimp. are different, give the ranges, otherwise (cr)
first & last residue of frag. 1: 39 49
first & last residue of frag. 2:
Molecule 3 has different length!
If the fragments for superimp. are different, give the ranges, otherwise (cr)
first & last residue of frag. 1: 3 13
first & last residue of frag. 2:
Molecule 4 has different length!
If the fragments for superimp. are different, give the ranges, otherwise (cr)
first & last residue of frag. 1: 3 13
first & last residue of frag. 2:
Atom order not checked, because of different molecular
lengthes or identifying
atoms to be displayed!
Atom order not checked, because of different molecular
lengthes or identifying
atoms to be displayed!
Atom order not checked, because of different molecular
lengthes or identifying
atoms to be displayed!
Atom order not checked, because of different molecular
lengthes or identifying
atoms to be displayed!
Atom order not checked, because of different molecular
lengthes or identifying
atoms to be displayed!
|
1=RMSD, 2=HBTAB, 3=VIOLAT, 4=ANGDIS, 5=DISPLAY, 6=READML,
7=SURFACE_NO_ATOM, 8=SURFACE_WITH_ATOM,
9=RADIUS_OF_GYRATION, 10=RINGSHIFT, 0=STOP: 0
STOP Oh, XAM! statement executed
|
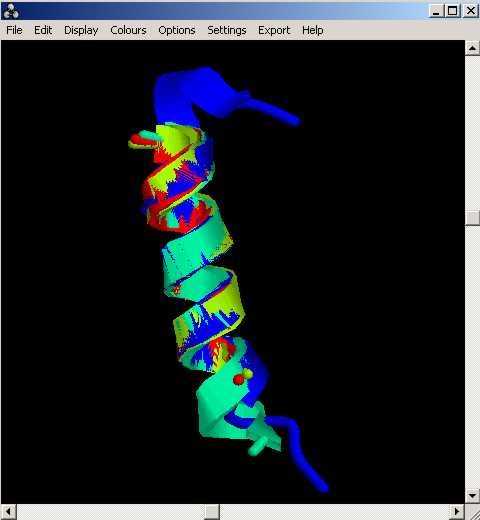
Un cop més, després dÇexecutar el programa, es generen dos arxius de sortida: totesquatre i totesquatre.pdb
Així doncs, el nombre més gran de residus que he pogut utilitzar per tal que el valor de RMSD sigui el més petit possible, ha estat de 11 residus. Els diferents valors de RMSD, entre les diferents hèlixs, es troba per sota del límit que es considera acceptable (< 2Å).
- Fes una comparació estructural de dos plegaments globin-like de la mateixa família de SCOP, amb el programa XAM.
Els passos que he seguit per tal dÇescollir les diferents proteïnes globin-like i després analitzar-les, ha estat:
- Anar a la pàgina dÇSCOP (SCOP!)
- Triar lÇopció de "Top of the Hierarchy"
- Triar "all alpha proteins"
- Triar "globin like"
- Triar Superfamília: Globin like
- Triar Família: Globins
- Escollir dÇentre les diferents espècies tant similars (Myoglobin,Hemoglobin ...)
Un cop he escollit les proteïnes de les diferents espècies amb què vull treballar, descarrego cada una de les diferents estructures en format .pdb (des de la pàgina de PDB, per exemple).
A continuació, sobre cada una dÇaquestes estructures triades, executo la comanda...:
$ perl /disc9/PERL/PDBtoSplitChain.pl -i nompdb.ent -o nompdb
... per tal de separar les diferents subcadenes que fomen cada una dÇaquestes estructures.
Finalment, les estructures que trio per a fer lÇaliniament estructural, són:
Human: 2mm1.ent
Whale: 1a6m.ent
Per a aquesta pràctica, realitzo tant un aliniament estructural (XAM), com un aliniament de seqüències (Clustalw). Com ja hem vist les diferents comandes per a lÇexecució de cadascun dels programes, directament, analitzarem els resultats.
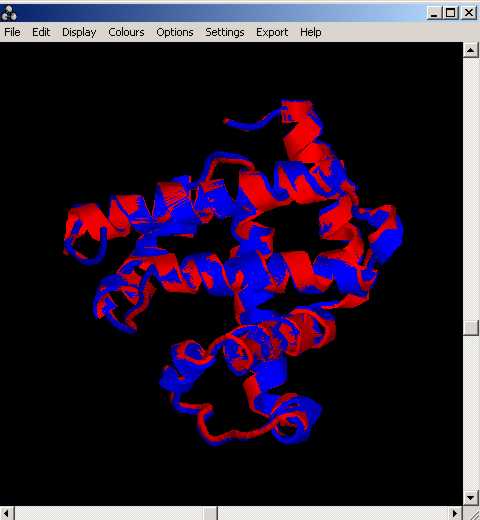
LÇexecuciµ de XAM, mostra dos arxius, human_whale i human_whale.pdb, mentre que de lÇexecuciµ de la comanda de Clustalw, nÇobtenim el següent aliniament: human_whale.aln a partir dÇun arxiu que conté les seqüències en format fasta (human_whale.fa)
El resultat obtingut és forÓa similar en ambdós casos: tant lÇaliniament estructural com el de seqüència ens estan indicant que les dues seqüències són molt semblants (lÇaliniament de Clustalw és molt bo) i que la seva estructura es conserva (lÇestructura se superposa quasi a la perfecció).
- DESCRIPCIÓ TEÒRICA DEL PROGRAMA
En aquesta part de la pràctica emprarem un programa dÇaliniament estructural (similar a XAM), però que en aquest cas utilitza lÇalgorisme dÇSmith-Waterman (SW) per tal de determinar lÇaliniament més ràpid existent entre les diferents estructures donades.
A diferència de lÇanterior programa, STAMP, no necessita lÇentrada dÇun interval de la seqüència a superposar, sinó que partint de les estructures (arxiu .pdb) nÇobté lÇaliniament global òptim.
- DESCRIPCIÓ PRÀCTICA DELS COMANDAMENTS DEL PROGRAMA I EXEMPLES DE COMANDAMENTS I CONDICIONS APLICADES EN LÇEXECUCIÓ
Un cop obtinguts els resultats de lÇaliniament estructural entre les diferents globines en lÇapartat 4.1, amb XAM, ara aplicarem un aliniament estructural amb el programa STAMP i tractarem de comparar els diferents resultats que se nÇobtenen.
Les diferents comandes utilitzades per tal dÇexecutar el programa, són:
[e14910.bio.acexs.au.upf@au48239 practica_4]$ cp /disc9/practica_4/STAMP/EXAMPLES/globin.tar .
Copiem la carpeta globin.tar en el nostre directori.
[e14910.bio.acexs.au.upf@au48239 practica_4]$ tar xvf globin
Desagrupem el directori .tar, per tal de poder-lo utilitzar.
[e14910.bio.acexs.au.upf@au48239 practica_4]$ cd globin
[e14910.bio.acexs.au.upf@au48239 practica_4]$ kwrite globin.domains
Editem un arxiu que presenti el format dÇentrada dels .pdb, tal i com ho mostra lÇarxiu de sortida adjunt.
[e14910.bio.acexs.au.upf@au48239 practica_4]$ stamp -l globin.domains -rough -n 2 -prefix globin > stampglobin
Amb aquesta comanda, fem correr el programa STAMP donant com arxiu dÇentrada: globin.domains. De lÇexecució del programa, resulten diferents arxius de sortida: globin.X (on X, és el nombre menys 1 de les seqüències emprades en lÇarxiu dÇentrada)
[e14910.bio.acexs.au.upf@au48239 practica_4]$ aconvertMod2.pl -in b -out c < globin.5 > aconvertglobin
Converteix un aliniament en format STAMP a un aliniament en format Clustalw. LÇopció -in b -out c, dennota el pas de BLOCK (format de sortida propi de lÇSTAMP) a CLUSTAL (format comú de lÇaliniament de seqüències). Aquesta comanda és la forma de mostrar lÇaliniament de sortida de lÇSTAMP en un format vertical.
[e14910.bio.acexs.au.upf@au48239 practica_4]$ transform -f globin.5 -g -o globin.5.pdb
Aquesta comanda transforma un arxiu de sortida dÇun programa determinat (dÇSTAMP, ALIGNFIT o SORTRANS) en un arxiu .pdb. LÇarxiu resultant contindrà totes les coordenades específiques transformades en un format definit. LÇopció -f, indica lÇarxiu a transformar; lÇopció -g, agrupa totes les coordenades transformades en un sol arxiu .pdb per tal de facilitar-ne la visualització en Rasmol; lÇopció -o, determina el nom de lÇarxiu de sortida.
[e14910.bio.acexs.au.upf@au48239 practica_4]$ rasmol globin.5.pdb
Visualització de la superposició resultant, tot utilitzant el programa Rasmol.
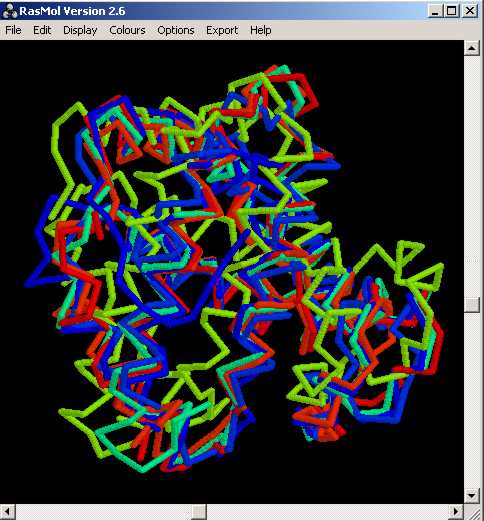
- RESULTATS OBTINGUTS DE LÇAPLICACIÓ DEL PROGRAMA ALS EXEMPLES COMENTATS I PROBLEMES PROPOSATS A LA PRÀCTICA
- Escull un plegament β-propeller i fes la superposició dels membres de la família, superfamília i el plegament.
Per tal dÇexecutar el programa STAMP, obtinc les diferents estructures a utilitzar a partir de lÇSCOP i de PDB.
Els passos que he seguit per tal dÇescollir les diferents proteïnes β-propeller like i després analitzar-les, ha estat:
- Anar a la pàgina dÇSCOP (SCOP!)
- Triar lÇopció de "Top of the Hierarchy"
- Triar "all beta proteins"
- Triar "7-bladed beta-propeller"
- Triar Superfamília: Quinoprotein amine dehydrogenase catalytic subunit i RCC1/BLIP-II
- Triar Família: per a la primera Superfamília, Methylamine dehydrogenase, H-chain i Quinohemoprotein amine dehydrogenase B chain i per a la segona Superfamília, Regulator of chromosome condensation RCC1 i of beta-lactamase inhibitor protein-II, BLIP-II.
- Escollir només dues espècies per a cada Família: 2mta i 1jju, per a la primera Família; 1a12 i 1jtd, per a la segona Família.
... ara lÇexecució del programa:
[e14910.bio.acexs.au.upf@au48239 practica_4]$ kwrite bpropeller.domains
[e14910.bio.acexs.au.upf@au48239 practica_4]$ stamp -l bpropeller.domains -rough -n 2 -prefix bpropeller > stampbpropeller
[e14910.bio.acexs.au.upf@au48239 practica_4]$ aconvertMod2.pl -in b -out c < bpropeller.3 > aconvertbpropeller
[e14910.bio.acexs.au.upf@au48239 practica_4]$ transform -f bpropeller.3 -g -o bpropeller.3.pdb
[e14910.bio.acexs.au.upf@au48239 practica_4]$ rasmol bpropeller.3.pdb
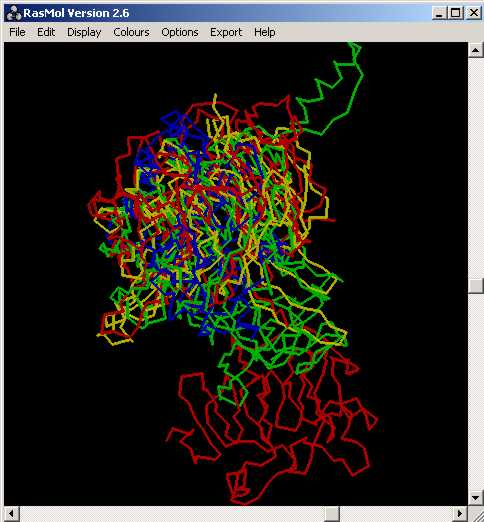
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}