- INTRODUCCIÓ A LA PRÀCTICA
Degut a que aquesta pràctica és forēa llarga i inclou l“execució de gran nombre de comandes així com de diferents programes, distribuiré la pràctica de la següent manera: en primer lloc, a mode d“introducció de les diferents comandes, utilitzaré els arxius fets a classe com a exemple.
Un cop executades les comandes fins a obtenir un model, passaré a comentar el que jo mateixa he obtingut.

- COMANDES BÀSIQUES EMPRADES I PROGRAMES UTILITZATS
Totes les comandes bàsiques utilitzades al llarg de la pràctica a mode de resum, són:
| PART 1: PSI-Blast i Clustalw |
En primer lloc, busco l“entrada corresponent a P11018 (subtilisin) a GenBank.
Edito un arxiu que contingui la seqüència en format fasta.
A continuació inicio la cerca en les diferents bases de dades (en aquest cas: PSI-Blast):
[e14910.bio.acexs.au.upf@au48239 practica5_1]$ /disc9/BLAST/EXE/blastpgp -i P11018.fa -d /disc9/DB/blast/swissprot -j 2 -C P11018.bls1 -o P11018.out
[e14910.bio.acexs.au.upf@au48239 practica5_1]$ /disc9/BLAST/EXE/blastpgp -i P11018.fa -d /disc9/DB/blast/pdb -j 2 -R P11018.bls1 -C P11018.bls2 -j 2 -o P11018.out2
Un cop executades les comandes anteriors, d“entre els resultats obtinguts, escullo de 2 a 8 seqüències homòlogues a la subtilisin. A continuació descarregaré l“estructura (format .pdb) de cada una d“elles així com la seqüència en format fasta.
A partir de les seqüències trobades en format fasta, creo un arxiu que les contingui totes (llista.fa)
Tot seguit, executo el programa d“aliniament de seqüències, Clustalw i el resultat que n“obtinc és: llista.aln.
En aquest apartat ens centrarem en l“aliniament estructural dels diferents arxius .pdb utilitzats en l“apartat anterior.
Després de l“aliniament estructural, crearem un perfil HMM a partir del paquet de programes continguts en HMMER.
Les diferents comandes utilitzades han estat:
[e14910.bio.acexs.au.upf@au48239 practica5_2]$ kwrite pract5_2.domains
[e14910.bio.acexs.au.upf@au48239 practica5_2]$ stamp -l pract5_2.domains -rough -n 2 -prefix pract5_2 > stamppract5_2
[e14910.bio.acexs.au.upf@au48239 practica5_2]$ aconvertMod2.pl -in b -out c < pract5_2.3 > pract5_2.3.aln
[e14910.bio.acexs.au.upf@au48239 practica5_2]$ transform -f pract5_2.3 -g -o pract5_2.3.pdb
[e14910.bio.acexs.au.upf@au48239 practica5_2]$ rasmol pract5_2.3.pdb
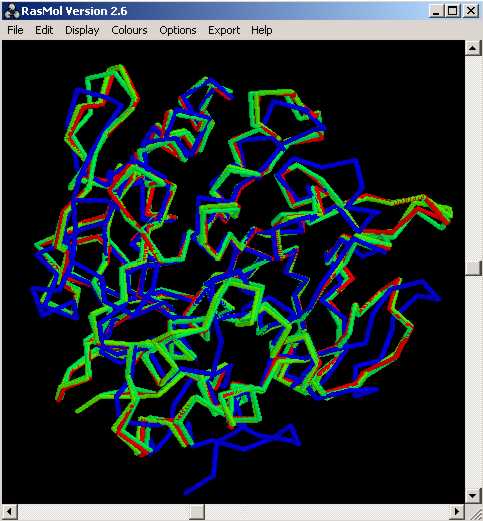
Ara realitzo modificacions sobre l“arxiu pract5_2.3.aln (li trec tot asterisc o caràcter rar) i el converteixo en: pract5_2.32.aln.
Un cop modificat l“arxiu, ja podré correr el programa per a la construcció d“HMM, hmmbuild.
[e14910.bio.acexs.au.upf@au48239 practica5_2]$ hmmbuild pract5_2.hmm pract5_2.32.aln
Ara creo un arxiu que contingui aquelles seqüències dels .pdb utilitzats per a fer l “STAMP i a més li afegirem la seqüència problema (P11018.fa), abans no inclosa entre la llista de .pdb (és obvi ja que la subtilisin no ha estat cristalitzada encara). L“arxiu resultant (alinia.fa), el correrem amb l“opció:
[e14910.bio.acexs.au.upf@au48239 practica5_2]$ hmmalign -o alinia.aln pract5_2.hmm alinia.fa
Ara, el que farem és construir un model a partir dels aliniaments de les pràctiques 5.1 (Clustalw) i 5.2 (hmmalign), amb el programa MODELLER.
Les comandes bàsiques utlitzades han estat:
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ PDBtoSplitChain.pl -i nomPDB.ent -o nomPDB
Comanda que separa les subcadenes contingudes en un arxiu .pdb i les guarda en arxius separats (per exemple una proteïna que té 4 cadenes i de l“execució d“aquesta comanda en resultaran quatre arxius: nompdbA, nompdbB, nompdbC i nompdbD)
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ aconvertMod2.pl -in c -out p < llista2.aln > llista2.ali
Aquesta comanda només és vàlida per als resultats obtinguts en l“apartat 5.1. L“opció -in c, fa referència al format introduït, Clustalw.
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ aconvertMod2.pl -in h -out p < alinia2.aln > alinia2.ali
Aquesta comanda només és vàlida per als resultats obtinguts en l“apartat 5.2. L“opció -in h, fa referència al format introduït, HMM.
Després de realitzar el passos anteriors, podem passar a configurar l“arxiu d“entrada (nomarxiu.top) al programa MODELLER. En aquest cas, ja que es tracta d“un exemple, deixarem l“arxiu tal qual i correrem el programa. Més tard sí el modificarem per a mostrar els models obtinguts.
Per a visualitzar l“arxiu:
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ kwrtie p7.top
I l“arxiu mostra...
# PRIMER: STEP 5
#
# This script should produce two models, 1fdx.B999901 and 1fdx.B999902.
#
# Before you run this script, do this: ln alignment.seg.ali fer2.ali
#
INCLUDE # Include the predefined TOP routines
SET ALNFILE = 'p7.ali' # alignment filename
SET KNOWNS = '1scjA' '1sbh' # codes of the templates
SET SEQUENCE = 'P11018' # code of the target
SET ATOM_FILES_DIRECTORY = './' # directories for input atom files
SET STARTING_MODEL= 1 # index of the first model
SET ENDING_MODEL = 2 # index of the last model
# (determines how many models to calculate)
SET DEVIATION = 2.0 # have to be >0 if more than 1 model
SET RAND_SEED = -12312 # to have different models from another TOP file
CALL ROUTINE = 'model' # do homology modelling
|
... i de l“execució del programa...:
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ mod p7.top
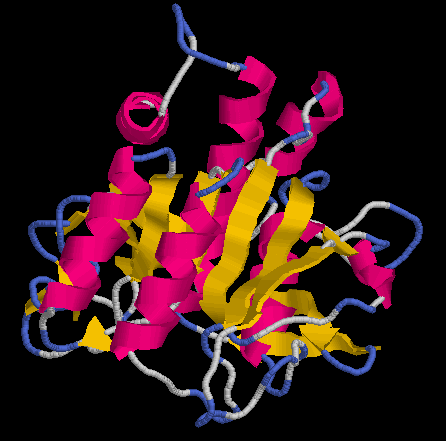
... en resulten diversos arxius d“entre els què se“n destaquen dos:
P11018.B99990001
P11018.B99990002
Val a dir que l“execució del programa MODELLER sempre genera els mateixos arxius de sortida (xxx.B99990001 i xxx.B99990002), així doncs és convenient modificar-ne el nom:
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ mv P11018.B99990001 P11018pdb1.ent
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ rasmol P11018pdb1.ent
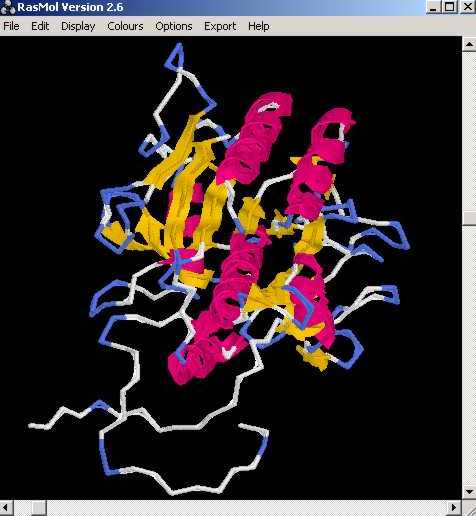
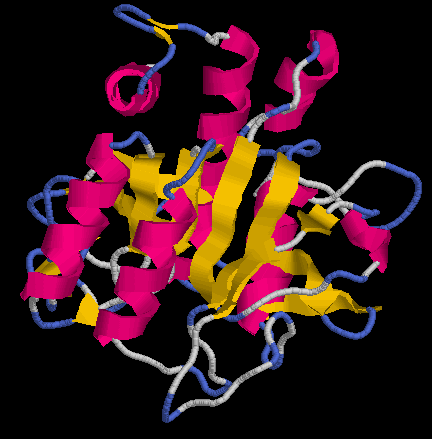
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ mv P11018.B99990002 P11018pdb2.ent
[e14910.bio.acexs.au.upf@au48239 practica5_3]$ rasmol P11018pdb2.ent
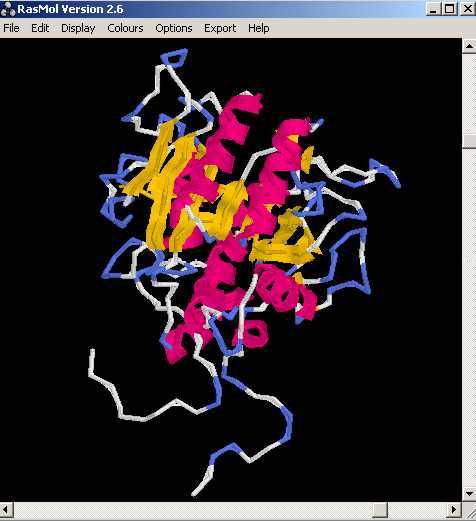
Un cop hem obtingut els models, podem passar a examinar la seva estructura...
L“execució de la comanda PROCHECK, genera molts arxius de sortida. Ara ens detindrem a comprovar què conté cadascun d“ells:
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ procheck_single nompdb.ent resolució
Per a cada model el nompdb.ent serà diferent (hi hem d“introduir el nom del model que haguem triat) i segons les estructures emprades en cada cas, haurem de modificar o no el valor de la resolució per tal d“executar la comanda PROCHECK.
La resolució d“una estructura es pot consultar tot revisant l“arxiu .pdb o .ent, des d“un editor de text.
... i els arxius de sortida:
| PROCHECK de P11018pdb1 i P11018pdb2 |
| Arxius de sortida |
| P11018pdb1 | P11018pdb2 | Descripció |
| Summary | Summary | P11018pdb1.sum i P11018pdb2.sum |
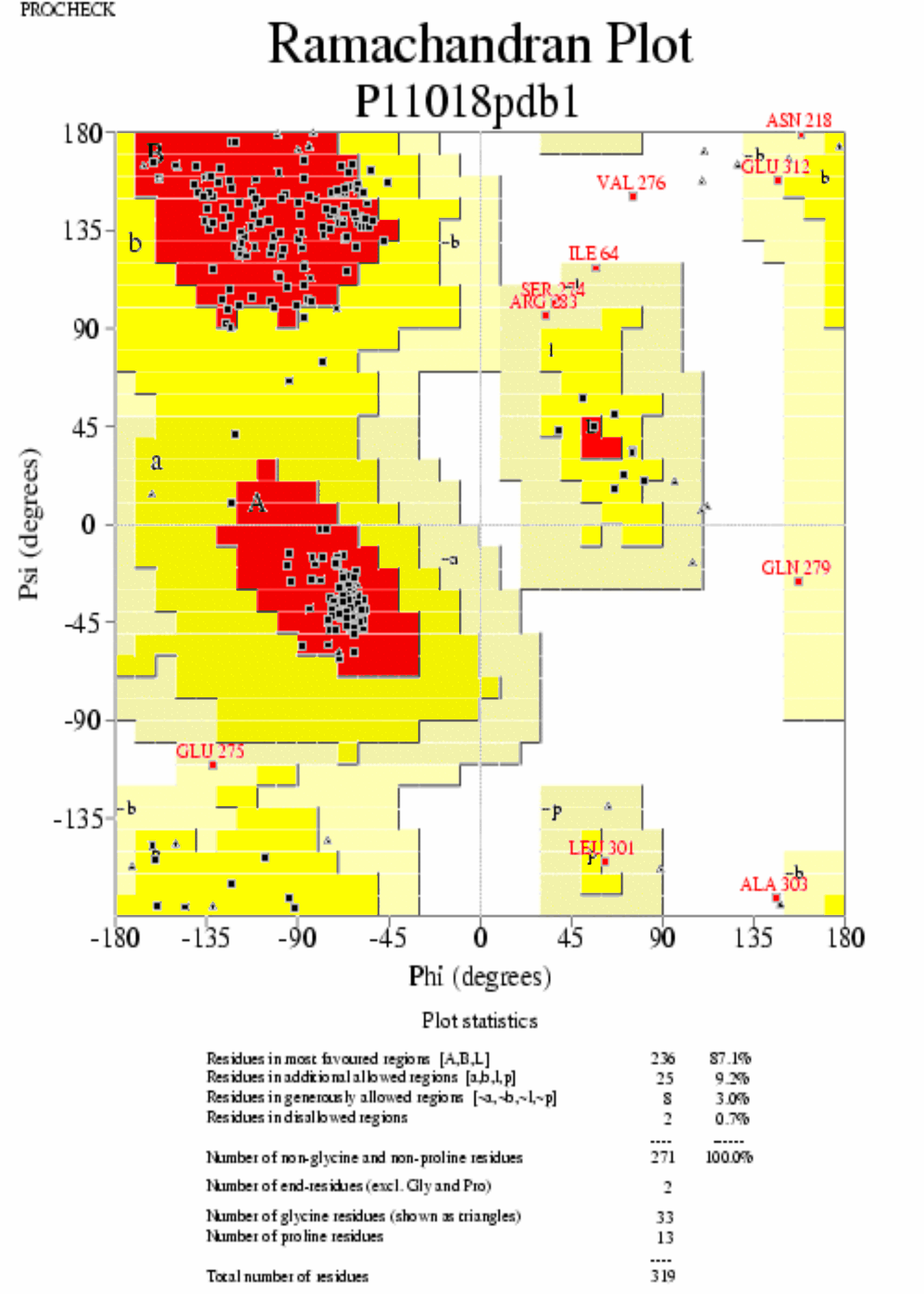 | 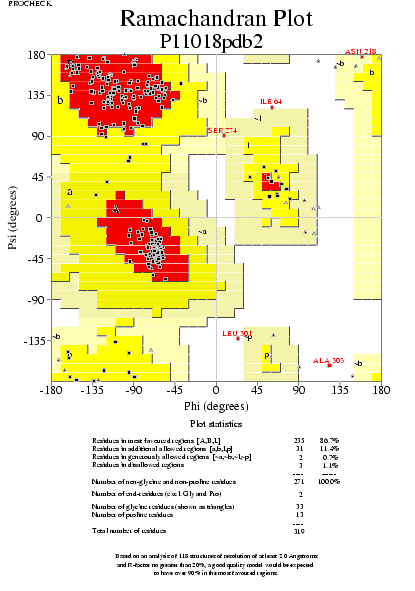 | P11018pdb1_01.ps i P11018pdb2_01.ps: Diagrama de Ramachandran per a tota l“estructura |
 | 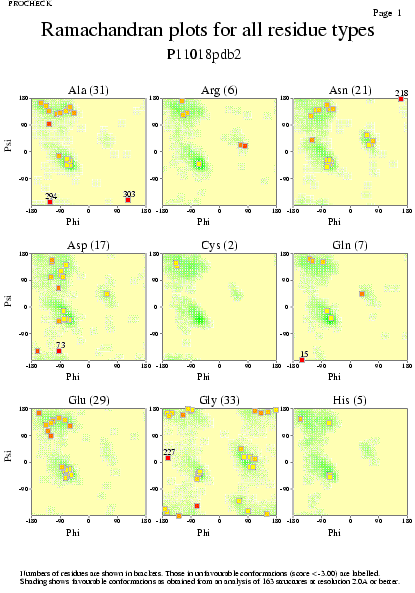 | P11018pdb1_02.ps i P11018pdb2_02.ps: Diagrama de Ramachandran per a cadasqun dels residus |
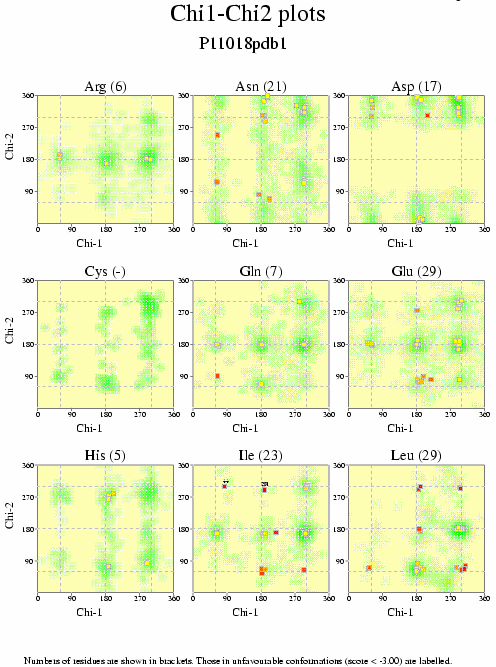 | 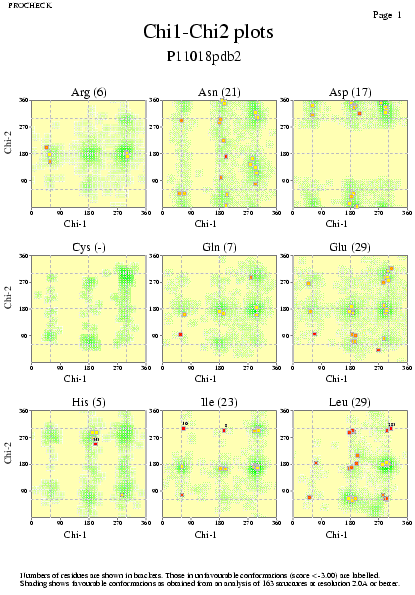 | P11018pdb1_03.ps i P11018pdb2_03.ps: Valors de Chi |
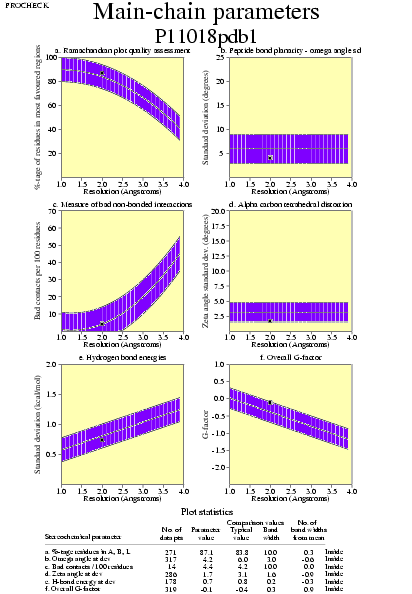 | 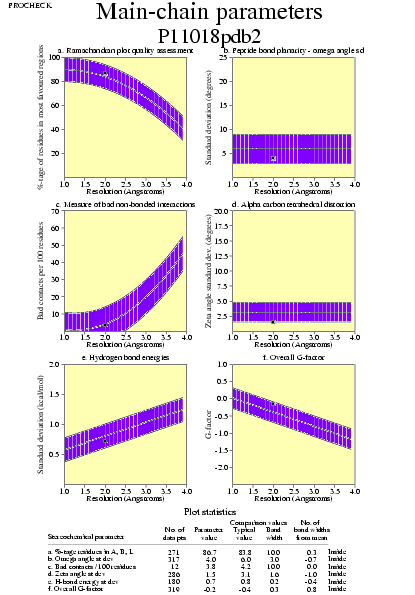 | P11018pdb1_04.ps i P11018pdb2_04.ps: Paràmetres de la cadena principal |
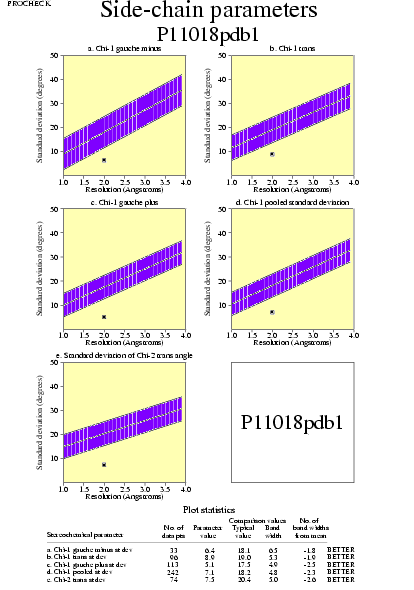 | 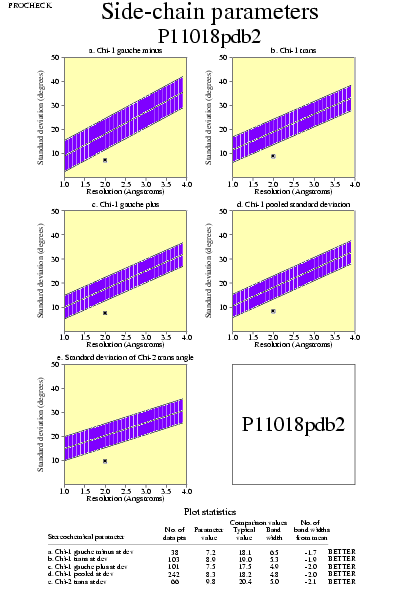 | P11018pdb1_05.ps i P11018pdb2_05.ps: Paràmetres de la cadena lateral |
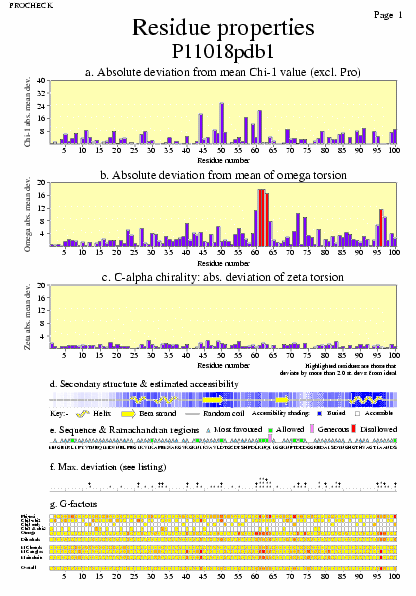 | 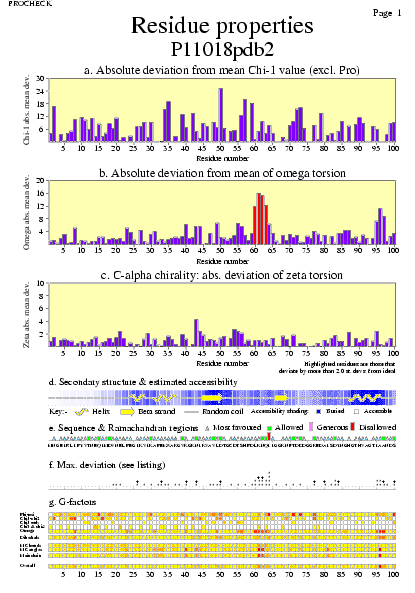 | P11018pdb1_06.ps i P11018pdb2_06.ps: Propietats del residus |
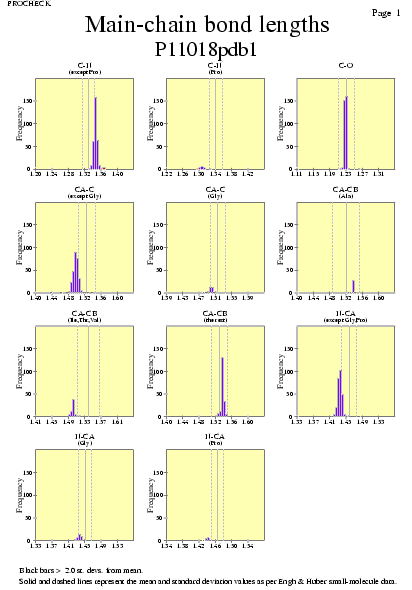 | 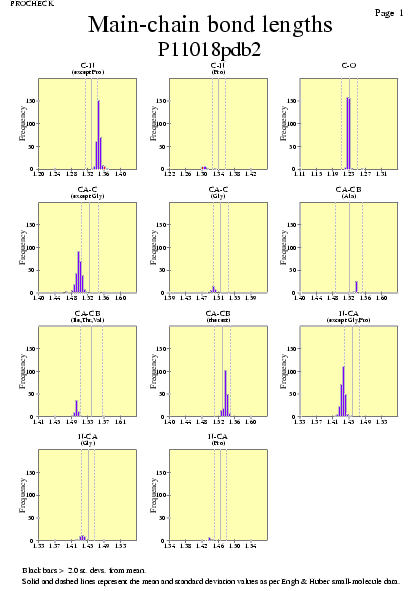 | P11018pdb1_07.ps i P11018pdb2_07.ps: Distància dels enllaēos en la cadena principal |
 |  | P11018pdb1_08.ps i P11018pdb2_08.ps: Angles dels enllaēos en la cadena principal |
 |  | P11018pdb1_09.ps i P11018pdb2_09.ps: Valors de RMS |
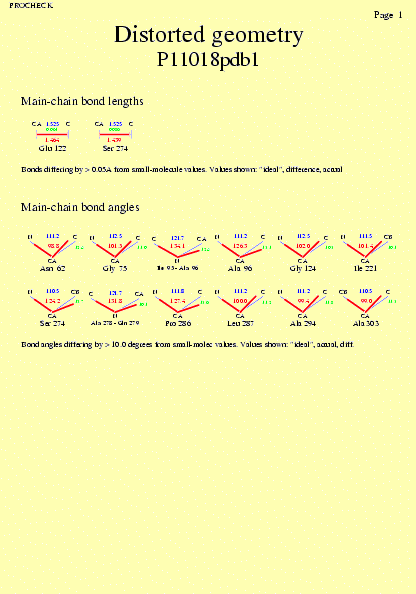 |  | P11018pdb1_10.ps i P11018pdb2_10.ps: Geometria de distorsió |
- MODELS EXPERIMENTALS OBTINGUTS
Un cop repassades les comandes amb el model de classe, ara procedirem a revisar el fet per mi...
Els meus models segueixen les mateixes comandes que el model anterior d“exemple, excepte a partir de l“execució del programa MODELLER. Mentre a classe vam usar un arxiu p7.top per a executar el programa tal qual, ara us mostro aquest arxiu modificat amb les estructures utilitzades per mi en ambdós models.
L“arxiu probl.top, l“equivalent al model de la pràctica 5.1 (Clustalw):
# PRIMER: STEP 5
#
# This script should produce two models, 1fdx.B999901 and 1fdx.B999902.
#
# Before you run this script, do this: ln alignment.seg.ali fer2.ali
#
INCLUDE # Include the predefined TOP routines
SET ALNFILE = 'llista2.ali' # alignment filename
SET KNOWNS = '1av7SEQ' '1pekSEQE' '1scjSEQA' # codes of the templates
SET SEQUENCE = 'P11018' # code of the target
SET ATOM_FILES_DIRECTORY = './' # directories for input atom files
SET STARTING_MODEL= 1 # index of the first model
SET ENDING_MODEL = 2 # index of the last model
# (determines how many models to calculate)
SET DEVIATION = 2.0 # have to be >0 if more than 1 model
SET RAND_SEED = -12312 # to have different models from another TOP file
CALL ROUTINE = 'model' # do homology modelling
|
... i l“arxiu probl2.top, corresponent al model obtingut per a la pràctica 5.2 (HMM):
# PRIMER: STEP 5
#
# This script should produce two models, 1fdx.B999901 and 1fdx.B999902.
#
# Before you run this script, do this: ln alignment.seg.ali fer2.ali
#
INCLUDE # Include the predefined TOP routines
SET ALNFILE = 'alinia2.ali' # alignment filename
SET KNOWNS = '1c9jSEQA' '1pekSEQE' # codes of the templates
SET SEQUENCE = 'P11018' # code of the target
SET ATOM_FILES_DIRECTORY = './' # directories for input atom files
SET STARTING_MODEL= 1 # index of the first model
SET ENDING_MODEL = 2 # index of the last model
# (determines how many models to calculate)
SET DEVIATION = 2.0 # have to be >0 if more than 1 model
SET RAND_SEED = -12312 # to have different models from another TOP file
CALL ROUTINE = 'model' # do homology modelling
|
Cal apreciar que per al primer model, he utilitzat tres templates (.pdb) i per al segon, només dos.
Això és: durant la pràctica vam tenir forēa dificultats per a executar el programa MODELLER degut a problemes amb les estructures, així que el nombre màxim de templates que he pogut fer servir (tot i haver-ho intentat forēa cops!) per a cada model, són aquests.
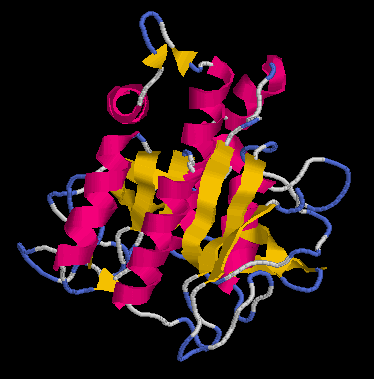
Els models que he obtingut de l“execució del programa MODELLER, han estat:
... per al primer model (pràctica 5.1: Clustalw):
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ mod probl.top
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ mv probl.B99990001 probl1amb3.ent
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ mv probl.B99990002 probl1amb3_2.ent
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ rasmol probl1amb3.ent
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ rasmol probl1amb3_2.ent
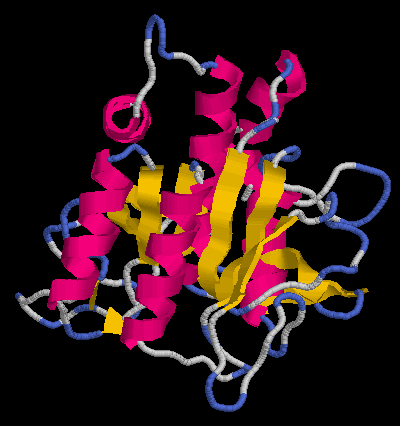
... i per al segon dels models (pràctica 5.2: HMM):
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ mod probl2.top
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ mv probl2.B99990001 probl2amb2.ent
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ mv probl2.B99990002 probl2amb2_2.ent
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ rasmol probl2amb2.ent
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ rasmol probl2amb2_2.ent
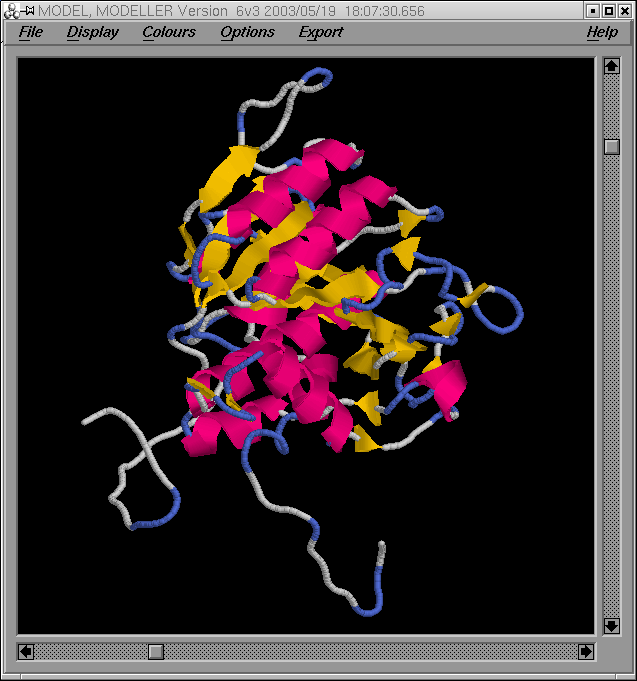
Un cop més, de l“execució de PROCHECK en resulten diversos arxius:
Per al model probl1amb3.ent i probl1amb3_2.ent:
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ procheck_single probl1amb3.ent 2.6
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ procheck_single probl1amb3_2.ent 2.6
| PROCHECK de probl1amb3 |
| Arxius de sortida |
| probl1amb3.ent | probl1amb3_2.ent | Descripció |
| Summary | Summary | probl1amb3.sum i probl1amb3_2.sum |
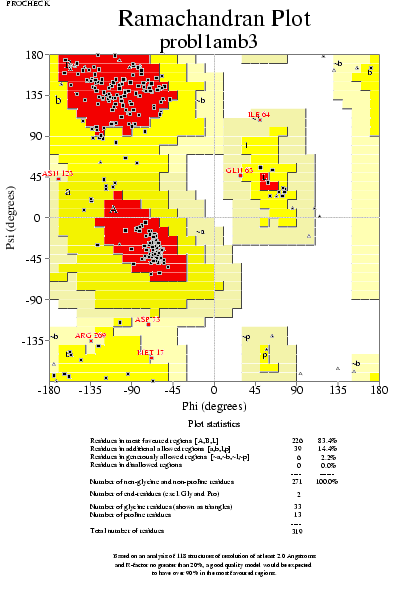 | 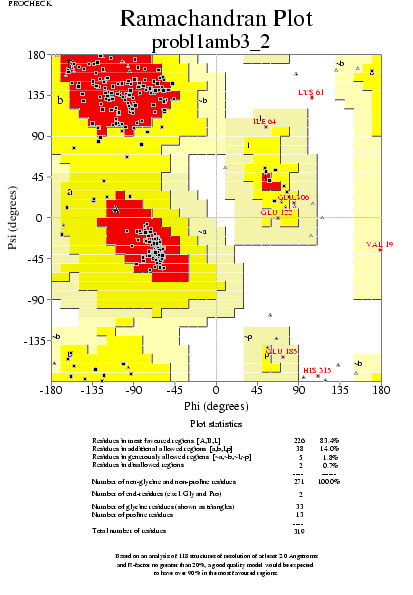 | probl1amb3_01.ps i probl1amb3_2_01.ps: Diagrama de Ramachandran per a tota l“estructura |
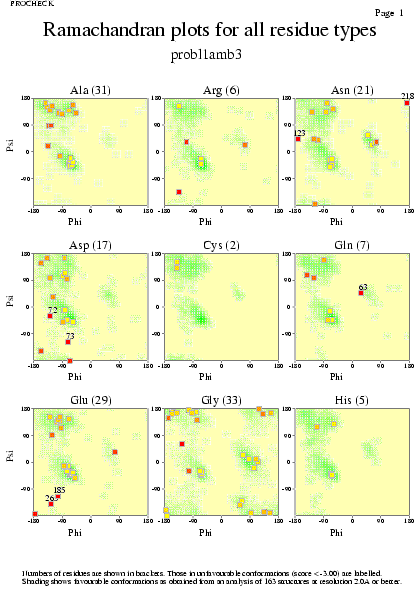 | 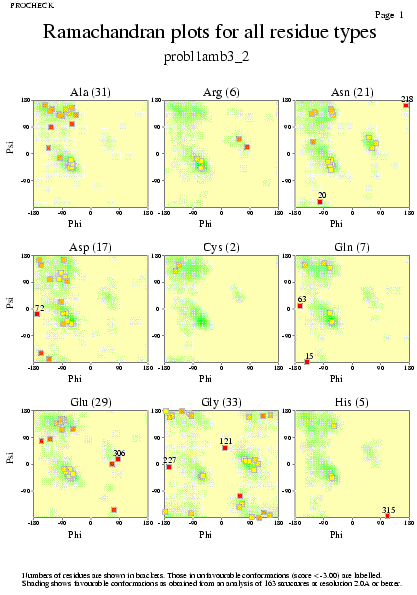 | probl1amb3_02.ps i probl1amb3_2_02.ps: Diagrama de Ramachandran per a cadasqun dels residus |
 |  | probl1amb3_03.ps i probl1amb3_2_03.ps: Valors de Chi |
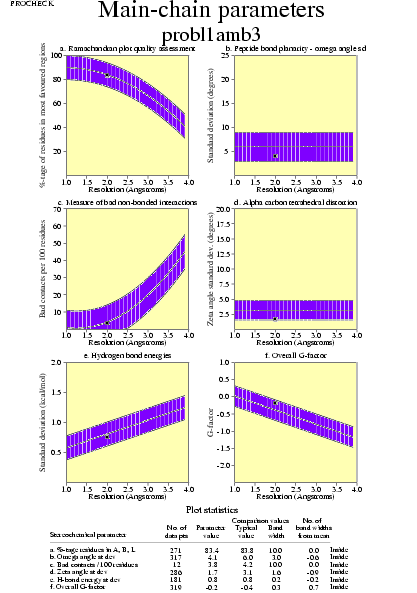 | 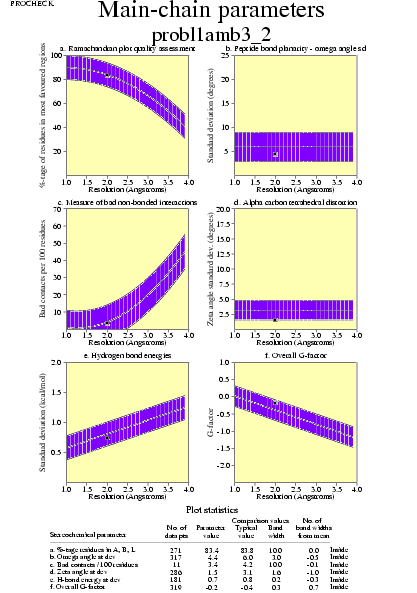 | probl1amb3_04.ps i probl1amb3_2_04.ps: Paràmetres de la cadena principal |
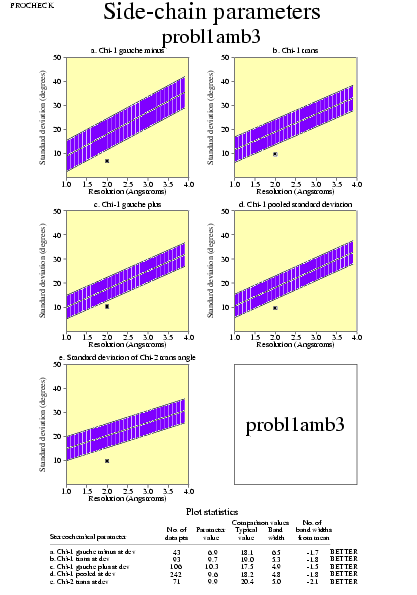 | 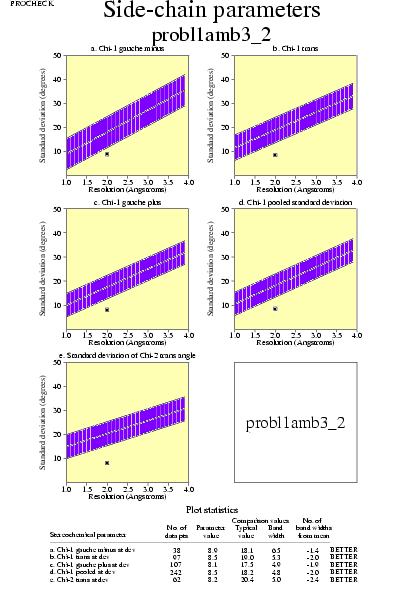 | probl1amb3_05.ps i probl1amb3_2_05.ps: Paràmetres de la cadena lateral |
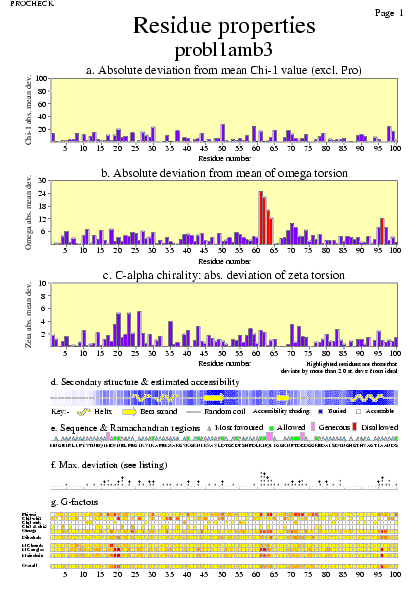 | 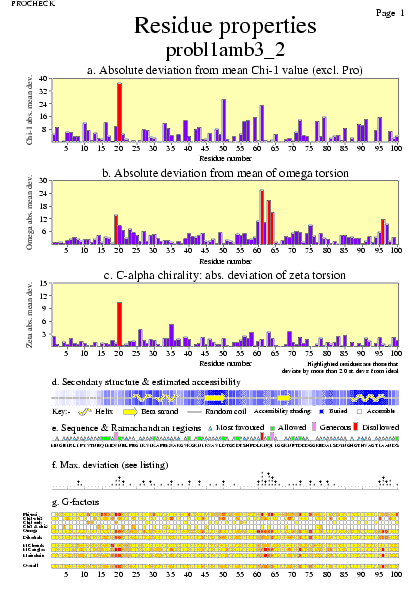 | probl1amb3_06.ps i probl1amb3_2_06.ps: Propietats del residus |
 |  | probl1amb3_07.ps i probl1amb3_2_07.ps: Distància dels enllaēos en la cadena principal |
 | 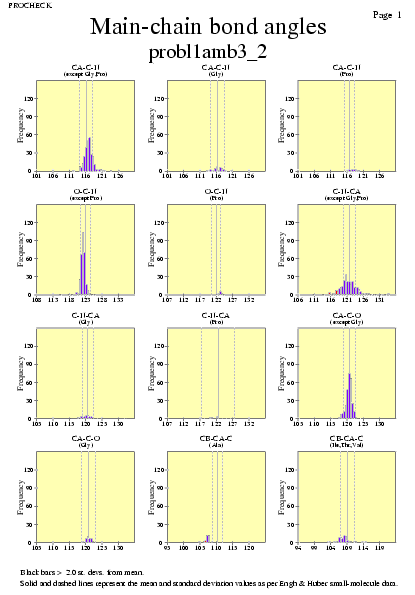 | probl1amb3_08.ps i probl1amb3_2_08.ps: Angles dels enllaēos en la cadena principal |
 | 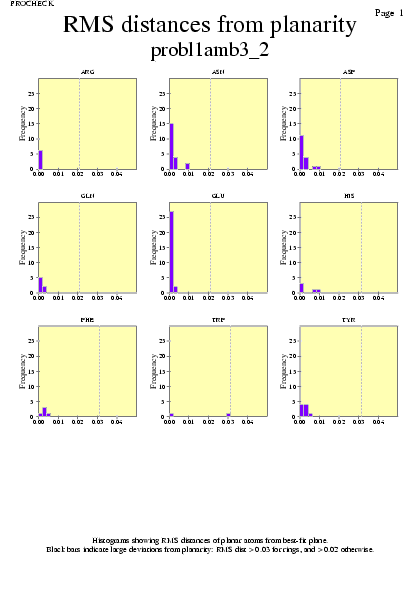 | probl1amb3_09.ps i probl1amb3_2_09.ps: Valors de RMS |
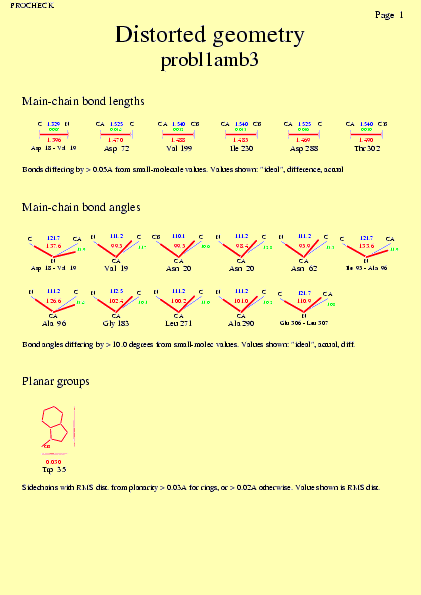 |  | probl1amb3_10.ps i probl1amb3_2_10.ps: Geometria de distorsió |
I per al model probl2amb2.ent i probl2amb2_2.ent:
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ procheck_single probl2amb2.ent 2.2
[e14910.bio.acexs.au.upf@au48239 practica5_4]$ procheck_single probl2amb2_2.ent 2.2
| PROCHECK de probl2amb2 |
| Arxius de sortida |
| probl2amb2.ent | probl2amb2_2.ent | Descripció |
| Summary | Summary | probl2amb2.sum i probl2amb2_2.sum |
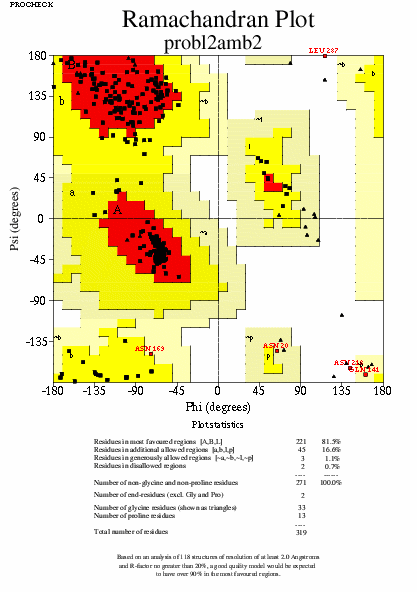 | 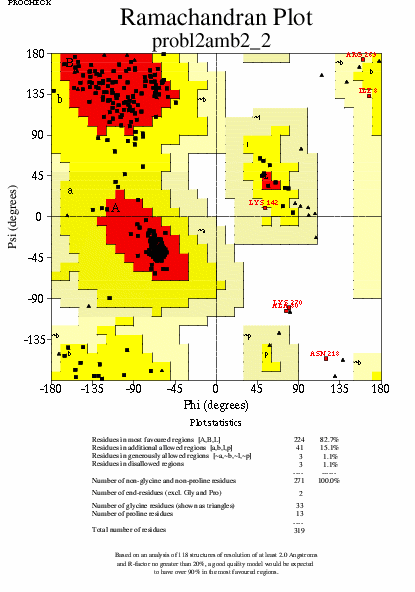 | probl2amb2_01.ps i probl2amb2_2_01.ps: Diagrama de Ramachandran per a tota l“estructura |
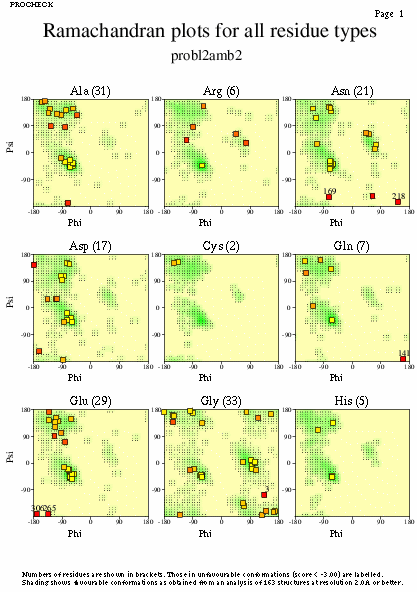 | 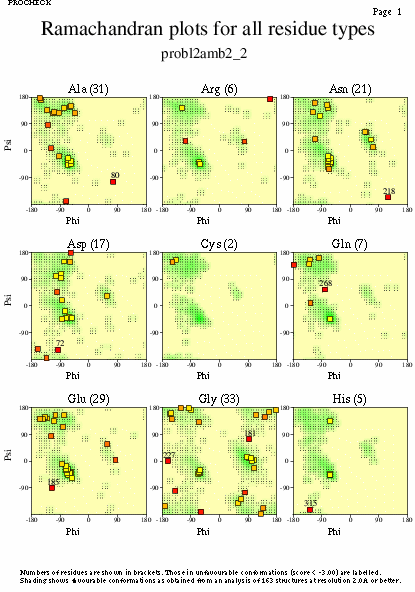 | probl2amb2_02.ps i probl2amb2_2_02.ps: Diagrama de Ramachandran per a cadasqun dels residus |
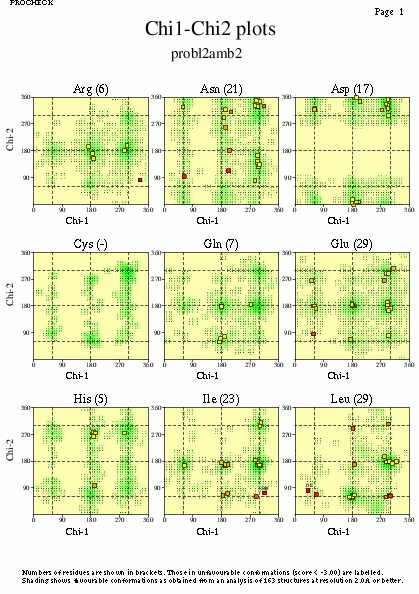 | 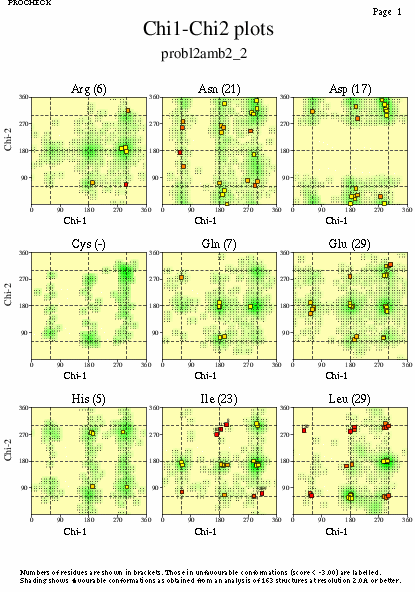 | probl2amb2_03.ps i probl2amb2_2_03.ps: Valors de Chi |
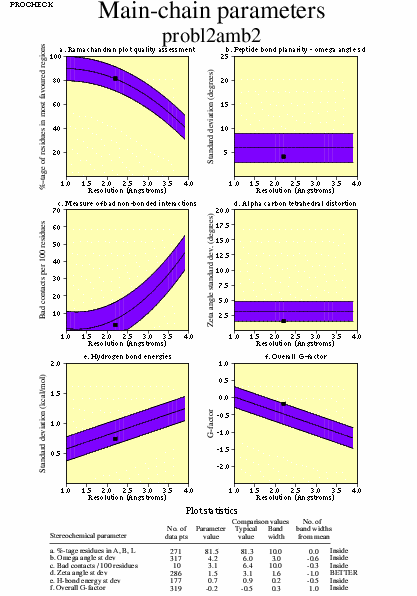 | 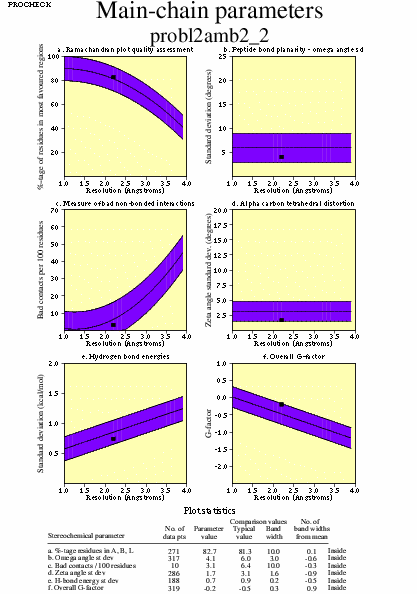 | probl2amb2_04.ps i probl2amb2_2_04.ps: Paràmetres de la cadena principal |
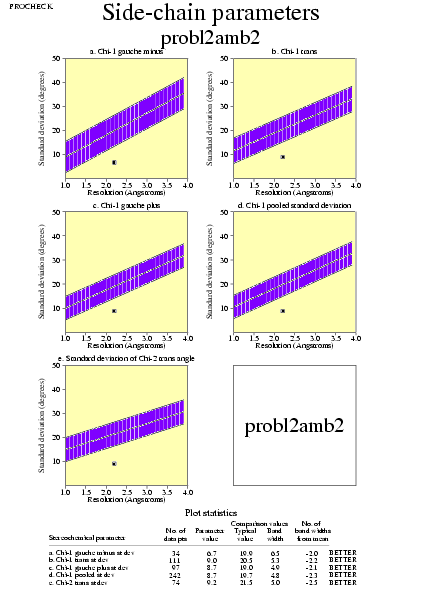 | 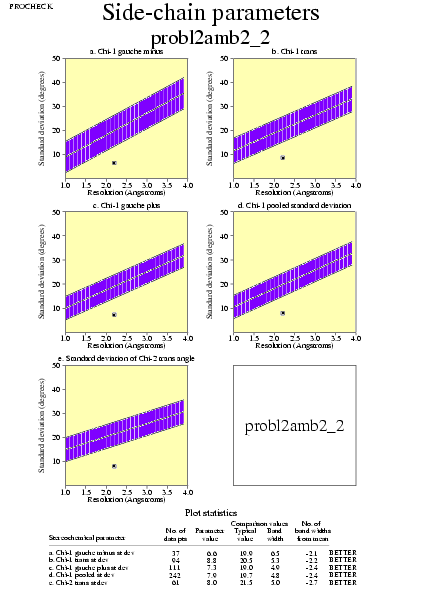 | probl2amb2_05.ps i probl2amb2_2_05.ps: Paràmetres de la cadena lateral |
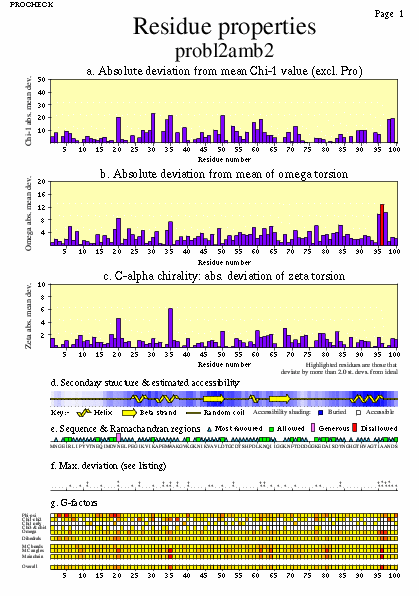 | 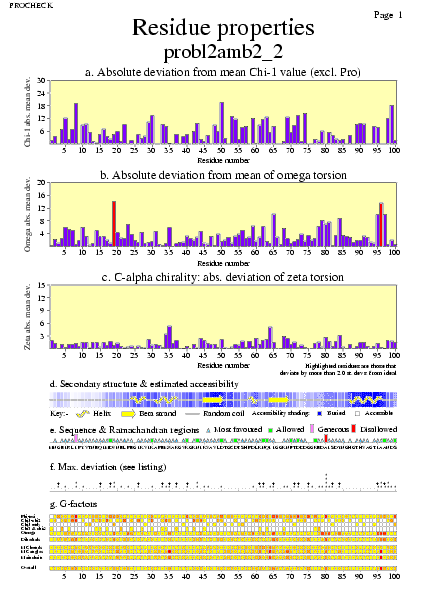 | probl2amb2_06.ps i probl2amb2_2_06.ps: Propietats del residus |
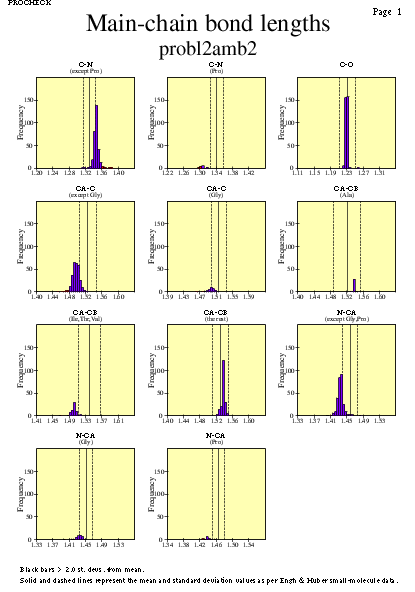 | 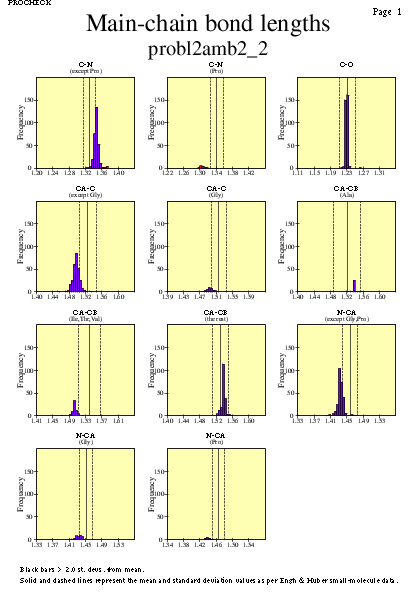 | probl2amb2_07.ps i probl2amb2_2_07.ps: Distància dels enllaēos en la cadena principal |
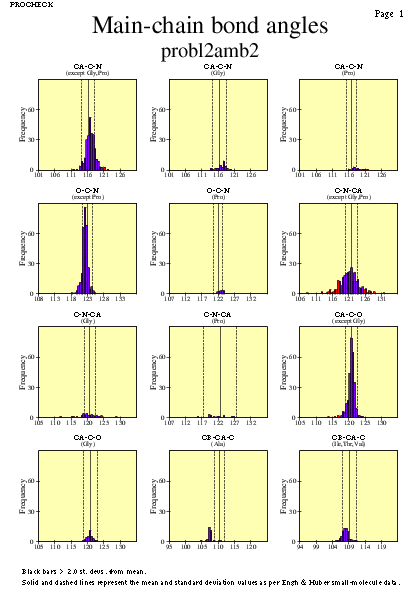 | 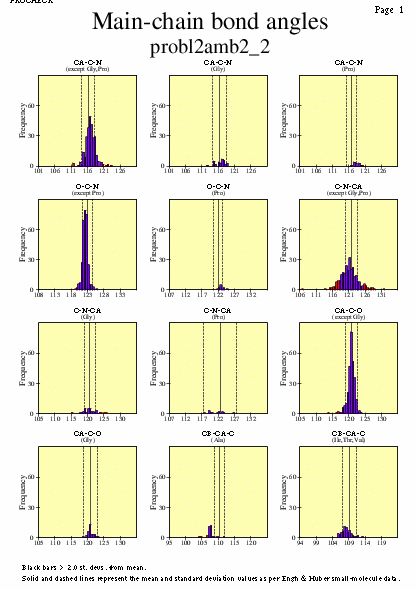 | probl2amb2_08.ps i probl2amb2_2_08.ps: Angles dels enllaēos en la cadena principal |
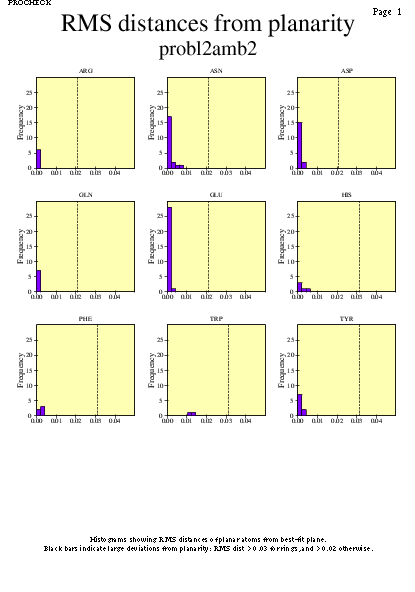 | 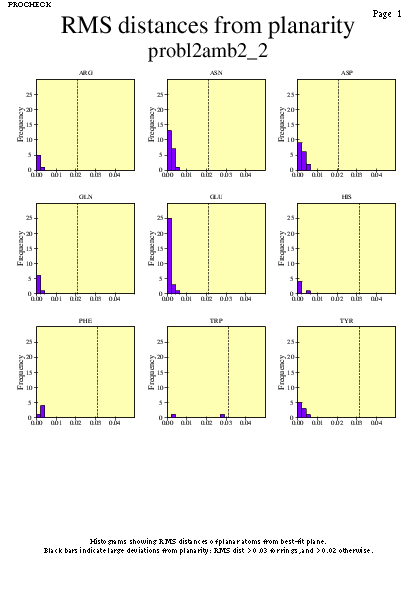 | probl2amb2_09.ps i probl2amb2_2_09.ps: Valors de RMS |
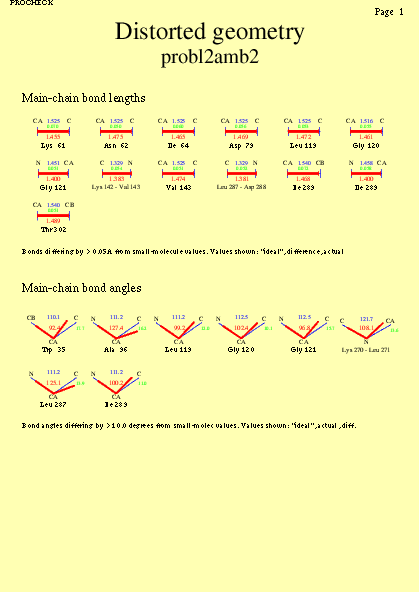 | 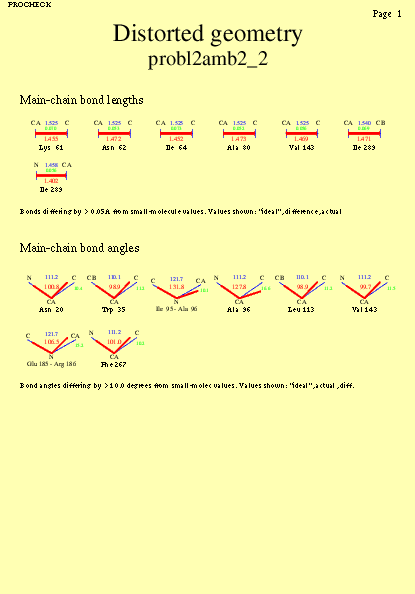 | probl2amb2_10.ps i probl2amb2_2_10.ps: Geometria de distorsió |
A continuació realitzarem una superposició dels quatre models obtinguts i tot seguit passarem a visualitzar-ne els resultats.
El programa que utilitzarem per tal de produir un aliniament estructural dels diferents models, serą XAM.
Val a dir que abans d“executar aquest programa i per tal d“obtenir un valor de RMSD fiable, hem retallat les cues de cada model, ja que aquestes zones acumulen un grau d“incertesa molt gran.
Aixķ doncs un cop tallades les cues, els models han passat a denominar-se:
- probl1amb3.ent: model1.pdb
- probl1amb3_2.ent: model2.pdb
- probl2amb2.ent: model3.pdb
- probl2amb2_2.ent: model4.pdb
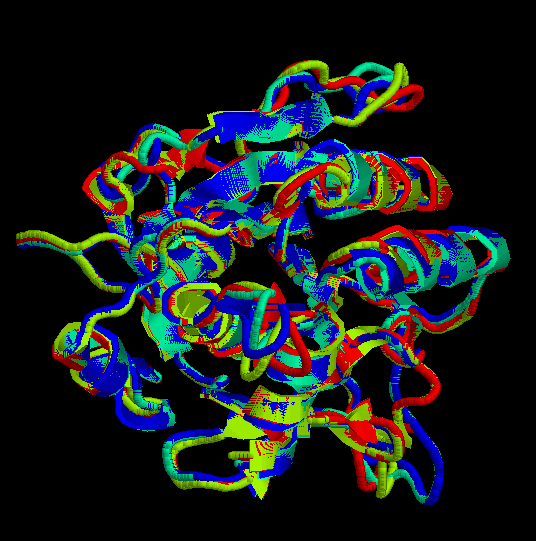
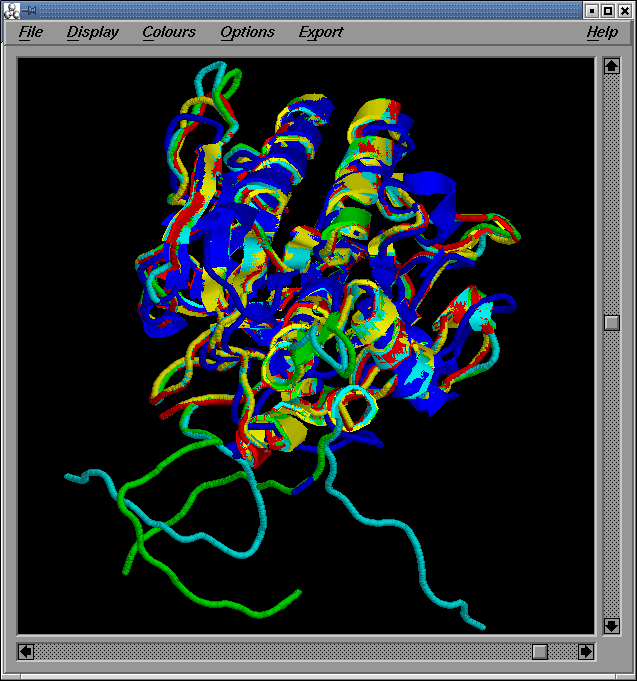
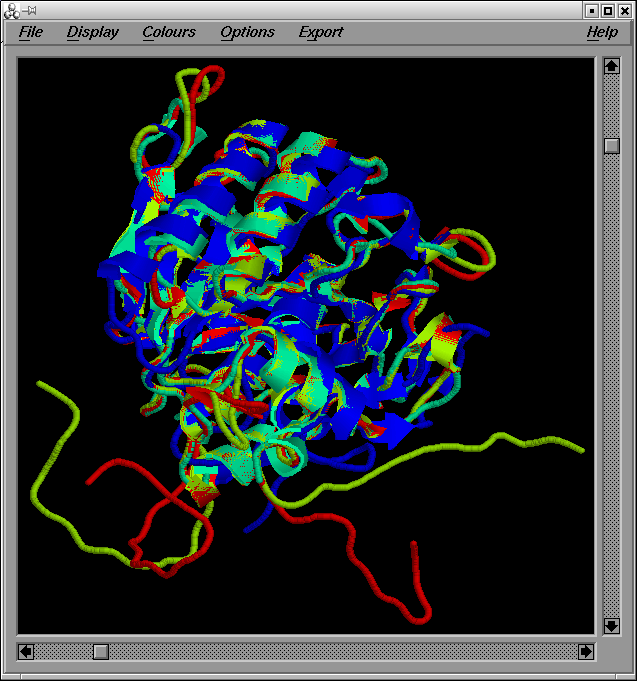
De l“execució del programa XAM, tot comparant els quatre models (model1.pdb, model2.pdb, model3.pdb i model4.pdb) els uns contra els altres i utilitzant tota la seqüčncia secera (un cop retallades les cues, resten 283 residus d“un total de 309), n“obtenim els resultats següents:
... i ja per a finalitzar, realitzarem una superposició dels diferents models amb els seus respectius templates (.pdb) emprats per a cadascun dels casos.
Per a realitzar aquest pas, emprarem el programa STAMP. I de l“execució d“aquest, en resulten els següents arxius:
...stamp... : outstampprobl1amb3 i outstampprobl2amb2
...aconvert... : outaconvertprobl1amb3 i outaconvertprobl2amb2
... i les estructures superposades visualment: probl1amb3.4.pdb i probl2amb2.3.pdb
Cal observar que de la comparació dels models amb els seus templates, es pot concluir que les estructures s'alinien estructuralment forēa bé exceptuant els loops, que són la regió més variable (ja ho hem pogut comprovar quan hem avaluat el valor de RMSD).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}