|
|
- DESCRIPCIÓ TEÒRICA DEL PROGRAMA
Grumos és l'interfície per a poder executar el programa GROMOS; un programa dissenyat per a realitzar la simulació de la dinàmica molecular per a estudis de sistemes biomoleculars.
Els objectius del programa, són:
- Predicció de la dependència de la conformació molecular amb l'entorn en el què es troba (aigua, etanol, cloroform, DMSO, solvent apolar, ...)
- Càlcul de les constants relatives d'enllaç tot evaluant diferències de l'energia lliure entre diversos complexos moleculars a partir d'integracions termodinàmiques, perturbacions i extrapolacions.
- Predicció de canvis energètics i estructurals causats per modificacionsd'aà en enzims o en parells de bases a l'ADN.
- Derivació en tres dimensions (3D) de l'estructura molecular basats en dades obtingudes a partir del NMR.
- Modelatge dinàmic de complexes moleculars
- Predicció de les propietats dels materials sota condicions extremes de temperatura i pressió, les quals poden ser experimentalment inaccessibles.

- DESCRIPCIÓ PRÀCTICA DELS COMANDAMENTS DEL PROGRAMA I EXEMPLES DE COMANDAMENTS I CONDICIONS APLICADES EN L´EXECUCIÓ
Realitzarem la simulació de la Dinàmica Molecular sense solvent, només en el millor dels models: probl1amb3_2.ent (model2.pdb).
Abans de correr el programa grumos, haurem de comprovar si aquesta estructura presenta residus de Cys prou aprop per a formar ponts disulfur.
Aquesta comprovació la podem fer tot visualitzant l'estructura en Rasmol i assenyalant-ne tots els residus de Cys:
RasMol> pick distance
RasMol>
Atom #1: CYS175.SG (1286)
RasMol>
Atom #2: CYS53.SG (405)
Distance CYS175.SG-CYS53.SG: 16.227
|
La distància entre el dos residus de Cys, és de 16.227Å, massa gran per a formar un pont disulfur.
Una altra cosa que cal revisar abans d'executar el programa, és comprovar la terminació del model en l'arxiu .pdb en el cas que haguem modificat l'estructura (si per exemple li hem tret les cues).
Ens hem d'assegurar que l'última fila de l'estructura presenti:
ATOM 2103 NH1 ARG 283 14.834 3.028 42.614 1.00198.16 1SG2104
ATOM 2104 NH2 ARG 283 16.193 1.104 42.428 1.00198.16 1SG2105
ATOM 2105 C ARG 283 13.218 5.282 46.378 1.00198.16 1SG2106
ATOM 2106 O ARG 283 14.115 6.046 46.732 1.00198.16 1SG2107
ATOM 2107 OXT ARG 283 12.720 5.305 45.134 1.00102.49 1SG2108
TER
|
Un cop revisats el possibles ponts S-S, comprovada la terminació del .pdb, només cal realitzar la següent comanda per tal de solucionar algun possible problema de format existent en l'estructura del model (consisteix a passar un filtre al nostre .pdb):
$ arrange.pl nommodel grumosnommodel.pdb
L'execució d'aquest interactiu programa mostra les següents instruccions per pantalla, de les quals en resalto:
[e14910.bio.acexs.au.upf@au48239 practica_7]$ grumos
This program prepares the drivers to run GROMOS package
You have to define the pathway in your directory
A Protein Data Bank (PDB) file is needed here
A generic name may be xxxx.pdb or whatever name you want
The only thing you have to comply with is the format PDB
Tell me path & file with the structure PDB :
NAME > /disc9/BE/e14910/practica_7/grumosmodel2.pdb
You have to select the name of a directory where you want to work
The default option is .../pro-grumos/
Give me the directory name : grumos-model2
mkdir: no se puede crear el directorio `grumos-model2': File exists
P R O - G R U M O S
Program to run GROMOS package, made in the
Institut de Biologia Fonamental (I.B.F.)
Universidad Autonoma de Barcelona. Spain
and
Department of Physical Chemistry
Uppsala University
Version : 2.0
Author: Baldomero Oliva Nov.1990
Revised and tested by : O. TAPIA Feb.1991
Last revision: Baldomero Oliva (Nov. 1993)
dl jun 2 10:13:32 CEST 2003
At this point you may be interested in carrying out
a free energy calculation, or/and dynamic run
you must define now the files where the restraints are defined
These files are necessary to run other inputs
Do you need some more files? yes/<no> : no
Do you want to clean the directory you gave me? yes/<no> : yes
Do you really want to clean that directory : grumos-model2 ? yes/<no> : yes
Now I am going to copy a set of driver names necessary to set up the run
cp /disc9/grumos93/interface/sh/driveropt_em /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to run the optimization
cp /disc9/grumos93/interface/sh/driveropt_emS /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to run the optimization of Complex Systems
cp /disc9/grumos93/interface/sh/driveropt_cem /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_cemS /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_cemw /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_cemi /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_cem /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_cemS /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_cemw /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_cemi /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_cem /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_top6 /disc9/BE/e14910/practica_7/grumos-model2
This are the drivers & programs to continue the
optimization
This is the program to choose the size of the BOX in
case you want to run a process with water molecules
This is the program to change the inputs when the size
of the BOX also change in case you want to run a process
with water molecules, because then also it can be removed
the radius of the boundaries and the radius to calculate
the interaction on the potential
cp /disc9/grumos93/interface/sh/driveropt_emw /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to run the optimization of one system
with water molecules
cp /disc9/grumos93/interface/sh/driveropt_emi /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to run the optimization of one system
with water molecules plus ions
This program reorganize the pdb file to run PROGROMOS
This program reorganize the water molecules of the
crystal as solvent molecules
cp /disc9/grumos93/interface/sh/exedriveropt_em /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to execute the optimization of the
system without water molecules and solvent simulated
cp /disc9/grumos93/interface/sh/exedriveropt_emS /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to execute the optimization of the
Complex Systems
cp /disc9/grumos93/interface/sh/exedriveropt_emw /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to execute driveropt_emw
cp /disc9/grumos93/interface/sh/exedriveropt_emi /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to execute driveropt_emi
I am sorry but I have to continue preparing all the drivers you will need
cp /disc9/grumos93/interface/sh/menu_grumos /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/menu_return /disc9/BE/e14910/practica_7/grumos-model2
This is the MENU of GRUMOS
cp /disc9/grumos93/interface/sh/menu_analyses /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/menu_returan /disc9/BE/e14910/practica_7/grumos-model2
This is the MENU for the analyses
cp /disc9/grumos93/interface/sh/driver_top3 /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_top5 /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_merge /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_reducmt /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_rmt /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_reducmt /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_patch /disc9/BE/e14910/practica_7/grumos-model2
/.driver_patch
cp /disc9/grumos93/interface/sh/exedriver_patch /disc9/BE/e14910/practica_7/grumos-model2
/.exedriver_patch
cp /disc9/grumos93/interface/sh/driver_unpatch /disc9/BE/e14910/practica_7/grumos-model2
/.driver_unpatch
cp /disc9/grumos93/interface/sh/exedriver_unpatch /disc9/BE/e14910/practica_7/grumos-model2
/.exedriver_unpatch
This are the drivers to run the topology of the system
cp /disc9/grumos93/interface/sh/driver_md2 /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_mdS /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_md2 /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_mdS /disc9/BE/e14910/practica_7/grumos-model2
This is the driver to run the DYNAMIC
cp /disc9/grumos93/interface/sh/driveropt_water /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_water /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_mergewater /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_mergeion /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_ion /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_ion /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_water /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_mergewater /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_mergeion /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_ion /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_water_untop /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driveropt_ion_untop /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_water_untop /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriveropt_ion_untop /disc9/BE/e14910/practica_7/grumos-model2
This are the drivers to add the water molecules and
the ions, and also to optimize the new coordinates
of the system
cp /disc9/grumos93/interface/sh/driver_cmd /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_cmdS /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_cmd /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_cmdS /disc9/BE/e14910/practica_7/grumos-model2
This are the drivers to continue the dynamic
cp /disc9/grumos93/interface/dat/cebador_em /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/exe/cont_md /disc9/BE/e14910/practica_7/grumos-model2
cp: no se puede abrir `/disc9/grumos93/interface/exe/cont_md' para lectura: Permission denied
cp /disc9/grumos93/interface/sh/cont_md2.f /disc9/BE/e14910/practica_7/grumos-model2
This are the drivers to make the loop to run the
dynamic and also the optimization
I am preparing all the drivers you will need
cp /disc9/grumos93/interface/sh/changps /disc9/BE/e14910/practica_7/grumos-model2
This are the programs to make the input of the DYNAMIC
I am preparing all the drivers you will need
This are the programs to make the input of the TOPOLOGY
I am preparing all the drivers you will need
This are the programs to make the input of the OPTIMIZATION
This are the the programs to chose the radius of cut-off
when there are water molecules in our system
I am preparing all the drivers you will need
cp /disc9/grumos93/interface/dat/nada /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/dat/dict37.dat /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/dat/dic37d.dat /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/dat/merge_top.dat /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/dat/make_box.dat /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/dat/spc216.dat /disc9/BE/e14910/practica_7/grumos-model2
This are the data files for the analyses and the
dynamic with water molecules.
The file WATER-EM.DAT_OLD can be fixed to make the
optimization of the waters and ions when surrounding
the protein.
DRIVERS AND PROGRAMS FOR THE ANALYSIS
cp /disc9/grumos93/interface/sh/exedriver_avx /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_avx /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_transform /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_transform /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_ajc /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_ajc /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_ahb /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_ahb /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_rms /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_rms /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_momentum /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_momentum /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_imsdm /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_imsdm /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_lsq /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/driver_lsq /disc9/BE/e14910/practica_7/grumos-model2
cp /disc9/grumos93/interface/sh/exedriver_comp /disc9/BE/e14910/practica_7/grumos-model2
R U N N I N G the P R O G R A M
Now you are invited to answer the questions of the menu_grumos
THERE IS NOT A TERMINAL IDENTIFICATION
IN YOUR DATA FILE
Check if the program runs, if it is not then follow
the next instructions.
You must include the finalization before to run
this program.
Write the oxygen terminal of the protein if it is not.
Include as a new line the same last one,substituting
the remark ATOM by TER in your PDB file and restart
the process again .
Options :
<a> =====> Continue
b =====> Break
a
Options :
<a> ====> Create INPUTS
b ====> RUN a process
c ====> ANALYSIS
d ====> NEW SIMULATION
e ====> Logout of this program
a
Options of INPUT :
<a> ====> TOPOLOGY
b ====> ENERGY OPTIMIZATION
c ====> DYNAMIC
d ====> Go to MENU
a
Options :
<a> =====> Single System
b =====> Complex System
c =====> Reduce and obtain the formatted topology
d =====> Modify topology
e =====> Go to MENU
a
T O P O L O G Y
This program read the PDB file. Then, it decides
if there are disulphide bridges by reading the number
of SSBOND with the residue CYS ( and not other!
e.g. CYSH ),it creates a GSF file (old WVG files)
and the input to run PROGMT.
The file PDB will be previously rebuilt, in order
to make the correct numbering of the residues, and
recalled as : NAME-OF-PROTEIN.pdbINP
It will be possible to change the name of the resi-
dues, but the PDB file will go on having the same
residue, that means that the only possibility for
changes will be between names which belong to the
same residue, for instance :
1) HISA can be changed into HISB
2) CYS can be changed into CYS1 or CYS2
3) GLU and ASP can be changed by GLUH and ASPH
etc.
It will be possible also to remove the prostetic
groups or other groups which apear in the crystal,
as for exemple water molecules and counter ions, and
these also will be removed when to make the GSF file
One title will be chosen to define all the stuff in
this directory. It would be interesting for your
own profit to write on it the name of the molecule,
the principal characteristic of the simulation, as
for instance the use or not of solvent, the radius
of cut-off and cut-on, the charge in the tails N
and C terminal, etc., and finally the date of the
work, but just remind that the title can not be
longer than 80 characters.
Finally it will be possible to choose between a si-
mulation with water molecules as solvent or with the
so called NIS ( non inertial solvent) model,and also
will be possible to choose the chemical state of the
tails depending on the pH.
In case the simulation with water molecules was selec-
ted, it will be also possible to choose the ionic
stength, and this will be obtained by adding sodium
cloride ions. Depending in the tolerance of the error
to get the ionic strength it will be made the BOX
which hold the system, that means , if the tolerance
is very small then the BOX will increase at the same
time than the number of ions until the correct point
will be reached.
T I T L E
grumosmodel2
Name of PDB file : grumosmodel2.pdb
Aminoacid( 1): GLY
Aminoacid( 2): ILE
Aminoacid( 3): LYSH
Aminoacid( 4): VAL
Aminoacid( 5): ILE
Aminoacid( 6): LYSH
Aminoacid( 7): ALA
Aminoacid( 8): PRO
Aminoacid( 9): GLU
Aminoacid( 10): MET
Aminoacid( 11): TRP
Aminoacid( 12): ALA
Aminoacid( 13): LYSH
Aminoacid( 14): GLY
Aminoacid( 15): VAL
Aminoacid( 16): LYSH
Aminoacid( 17): GLY
Aminoacid( 18): LYSH
Aminoacid( 19): ASN
Aminoacid( 20): ILE
Aminoacid( 21): LYSH
Aminoacid( 22): VAL
Aminoacid( 23): ALA
Aminoacid( 24): VAL
Aminoacid( 25): LEU
Aminoacid( 26): ASP
Aminoacid( 27): THR
Aminoacid( 28): GLY
Aminoacid( 29): CYSH
Aminoacid( 30): ASP
Aminoacid( 31): THR
Aminoacid( 32): SER
Aminoacid( 33): HISA
Aminoacid( 34): PRO
Aminoacid( 35): ASP
Aminoacid( 36): LEU
Aminoacid( 37): LYSH
Aminoacid( 38): ASN
Aminoacid( 39): GLN
Aminoacid( 40): ILE
Aminoacid( 41): ILE
Aminoacid( 42): GLY
Aminoacid( 43): GLY
Aminoacid( 44): LYSH
Aminoacid( 45): ASN
Aminoacid( 46): PHE
Aminoacid( 47): THR
Aminoacid( 48): ASP
Aminoacid( 49): ASP
Aminoacid( 50): ASP
Aminoacid( 51): GLY
Aminoacid( 52): GLY
Aminoacid( 53): LYSH
Aminoacid( 54): GLU
Aminoacid( 55): ASP
Aminoacid( 56): ALA
Aminoacid( 57): ILE
Aminoacid( 58): SER
Aminoacid( 59): ASP
Aminoacid( 60): TYR
Aminoacid( 61): ASN
Aminoacid( 62): GLY
Aminoacid( 63): HISA
Aminoacid( 64): GLY
Aminoacid( 65): THR
Aminoacid( 66): HISA
Aminoacid( 67): VAL
Aminoacid( 68): ALA
Aminoacid( 69): GLY
Aminoacid( 70): THR
Aminoacid( 71): ILE
Aminoacid( 72): ALA
Aminoacid( 73): ALA
Aminoacid( 74): ASN
Aminoacid( 75): ASP
Aminoacid( 76): SER
Aminoacid( 77): ASN
Aminoacid( 78): GLY
Aminoacid( 79): GLY
Aminoacid( 80): ILE
Aminoacid( 81): ALA
Aminoacid( 82): GLY
Aminoacid( 83): VAL
Aminoacid( 84): ALA
Aminoacid( 85): PRO
Aminoacid( 86): GLU
Aminoacid( 87): ALA
Aminoacid( 88): SER
Aminoacid( 89): LEU
Aminoacid( 90): LEU
Aminoacid( 91): ILE
Aminoacid( 92): VAL
Aminoacid( 93): LYSH
Aminoacid( 94): VAL
Aminoacid( 95): LEU
Aminoacid( 96): GLY
Aminoacid( 97): GLY
Aminoacid( 98): GLU
Aminoacid( 99): ASN
Aminoacid(100): GLY
Aminoacid(101): SER
Aminoacid(102): GLY
Aminoacid(103): GLN
Aminoacid(104): TYR
Aminoacid(105): GLU
Aminoacid(106): TRP
Aminoacid(107): ILE
Aminoacid(108): ILE
Aminoacid(109): ASN
Aminoacid(110): GLY
Aminoacid(111): ILE
Aminoacid(112): ASN
Aminoacid(113): TYR
Aminoacid(114): ALA
Aminoacid(115): VAL
Aminoacid(116): GLU
Aminoacid(117): GLN
Aminoacid(118): LYSH
Aminoacid(119): VAL
Aminoacid(120): ASP
Aminoacid(121): ILE
Aminoacid(122): ILE
Aminoacid(123): SER
Aminoacid(124): MET
Aminoacid(125): SER
Aminoacid(126): LEU
Aminoacid(127): GLY
Aminoacid(128): GLY
Aminoacid(129): PRO
Aminoacid(130): SER
Aminoacid(131): ASP
Aminoacid(132): VAL
Aminoacid(133): PRO
Aminoacid(134): GLU
Aminoacid(135): LEU
Aminoacid(136): LYSH
Aminoacid(137): GLU
Aminoacid(138): ALA
Aminoacid(139): VAL
Aminoacid(140): LYSH
Aminoacid(141): ASN
Aminoacid(142): ALA
Aminoacid(143): VAL
Aminoacid(144): LYSH
Aminoacid(145): ASN
Aminoacid(146): GLY
Aminoacid(147): VAL
Aminoacid(148): LEU
Aminoacid(149): VAL
Aminoacid(150): VAL
Aminoacid(151): CYSH
Aminoacid(152): ALA
Aminoacid(153): ALA
Aminoacid(154): GLY
Aminoacid(155): ASN
Aminoacid(156): GLU
Aminoacid(157): GLY
Aminoacid(158): ASP
Aminoacid(159): GLY
Aminoacid(160): ASP
Aminoacid(161): GLU
Aminoacid(162): ARG
Aminoacid(163): THR
Aminoacid(164): GLU
Aminoacid(165): GLU
Aminoacid(166): LEU
Aminoacid(167): SER
Aminoacid(168): TYR
Aminoacid(169): PRO
Aminoacid(170): ALA
Aminoacid(171): ALA
Aminoacid(172): TYR
Aminoacid(173): ASN
Aminoacid(174): GLU
Aminoacid(175): VAL
Aminoacid(176): ILE
Aminoacid(177): ALA
Aminoacid(178): VAL
Aminoacid(179): GLY
Aminoacid(180): SER
Aminoacid(181): VAL
Aminoacid(182): SER
Aminoacid(183): VAL
Aminoacid(184): ALA
Aminoacid(185): ARG
Aminoacid(186): GLU
Aminoacid(187): LEU
Aminoacid(188): SER
Aminoacid(189): GLU
Aminoacid(190): PHE
Aminoacid(191): SER
Aminoacid(192): ASN
Aminoacid(193): ALA
Aminoacid(194): ASN
Aminoacid(195): LYSH
Aminoacid(196): GLU
Aminoacid(197): ILE
Aminoacid(198): ASP
Aminoacid(199): LEU
Aminoacid(200): VAL
Aminoacid(201): ALA
Aminoacid(202): PRO
Aminoacid(203): GLY
Aminoacid(204): GLU
Aminoacid(205): ASN
Aminoacid(206): ILE
Aminoacid(207): LEU
Aminoacid(208): SER
Aminoacid(209): THR
Aminoacid(210): LEU
Aminoacid(211): PRO
Aminoacid(212): ASN
Aminoacid(213): LYSH
Aminoacid(214): LYSH
Aminoacid(215): TYR
Aminoacid(216): GLY
Aminoacid(217): LYSH
Aminoacid(218): LEU
Aminoacid(219): THR
Aminoacid(220): GLY
Aminoacid(221): THR
Aminoacid(222): SER
Aminoacid(223): MET
Aminoacid(224): ALA
Aminoacid(225): ALA
Aminoacid(226): PRO
Aminoacid(227): HISA
Aminoacid(228): VAL
Aminoacid(229): SER
Aminoacid(230): GLY
Aminoacid(231): ALA
Aminoacid(232): LEU
Aminoacid(233): ALA
Aminoacid(234): LEU
Aminoacid(235): ILE
Aminoacid(236): LYSH
Aminoacid(237): SER
Aminoacid(238): TYR
Aminoacid(239): GLU
Aminoacid(240): GLU
Aminoacid(241): GLU
Aminoacid(242): SER
Aminoacid(243): PHE
Aminoacid(244): GLN
Aminoacid(245): ARG
Aminoacid(246): LYSH
Aminoacid(247): LEU
Aminoacid(248): SER
Aminoacid(249): GLU
Aminoacid(250): SER
Aminoacid(251): GLU
Aminoacid(252): VAL
Aminoacid(253): PHE
Aminoacid(254): ALA
Aminoacid(255): GLN
Aminoacid(256): LEU
Aminoacid(257): ILE
Aminoacid(258): ARG
Aminoacid(259): ARG
Number of aA in the chain: 259
Do you want change the name of some residue, YES/<NO> : NO
CYS residues list :
Residue CYS : 29
Residue CYS : 151
You must call CYS1-CYS2 the residues with the bond
Are there disulphide bridges? YES/<NO> : NO
Do you want change the name of some residue, YES/<NO> : NO
Options
<1> Non inertial solvent, NIS model
2 Explicit WATER SPC simulation
3 Explicit WATER SPC/E simulation
4 Charged Force Field in vacuum
Value: 1
Options
<1> Head NH3 and Tail COO ==> 6 ( pH ) 8
2 Head NH3 and Tail COOH ==> 0 ( pH ) 2
3 Head NH2 and Tail COO ==> 12 ( pH ) 14
4 Head NH2 and Tail COOH ==> Specific cases
Value: 1
Options :
<a> ====> Create INPUTS
b ====> RUN a process
c ====> ANALYSIS
d ====> NEW SIMULATION
e ====> Logout of this program
b
Options to RUN :
<a> ====> TOPOLOGY
b ====> ENERGY OPTIMIZATION
c ====> DYNAMIC
d ====> Go to MENU
a
Options :
<a> =====> Single System
b =====> Complex System
c =====> Reduce and obtain the formatted topology
d =====> Modify topology
e =====> Transform formated to binary topology
f =====> Modify the water BOX
g =====> Add water molecules to the optimized solute
h =====> Add counter ions to the solvated solute
i =====> Go to MENU
a
Directory where is the work :
/disc9/BE/e14910/practica_7/grumos-model2
Running the topology without ions
rm: no se puede borrar `fort.10': No such file or directory
rm: no se puede borrar `fort.11': No such file or directory
rm: no se puede borrar `fort.12': No such file or directory
rm: no se puede borrar `fort.9': No such file or directory
ERRORS IN THE OUTPUT OF PRUGMT :
LAST 5 LINES OF PRUGMT-OUTPUT
NAEX14: 4069
NATOM NCAG NAEX NAEX1-4 NBONH NBON NTHEH NTHE
2403 1464 6332 4069 491 1939 912 2623
NQHIH NQHI NPHIH NPHI NRPD NRPA
400 765 91 1230 0 0
The topology is ready
Including polar Hydrogens
491 HYDROGEN ATOMS WILL BE GENERATED
ERRORS IN THE OUTPUT OF PRUGCH :
LAST 5 LINES OF PRUGCH-OUTPUT
3. A T O M C O O R D I N A T E S :
grumosmodel2
Options :
<a> ====> Create INPUTS
b ====> RUN a process
c ====> ANALYSIS
d ====> NEW SIMULATION
e ====> Logout of this program
a
Options of INPUT :
<a> ====> TOPOLOGY
b ====> ENERGY OPTIMIZATION
c ====> DYNAMIC
d ====> Go to MENU
b
Have you run the TOPOLOGY program ? no/<yes> yes
Options :
<a> =====> Single System
b =====> Complex System
a
O P T I M I Z A T I O N
The optimization can be done by one of three
different geometry methods :
Steepest Descent
Conjugate Gradient
Both methods used alternatively
The program PROEM will be run several times, and
also this will be chosen.
Each run involve several steps, and each step
involves a value called LAMBDA to obtain a new
set of coordinates. It will be necessary to define
the maximum value of LAMBDA and the first value to
start the optimization.
The number of steps geting a new set of coordinates
will be also chosen for each run.
In case the method of Conjugate Gradient was chosen,
it will be necessary to especify how many times the
energy gradient in the step must be modified
Options
<1> Conjugate Gradient & Steepest Descent
2 Steepest Descent
3 Conjugate Gradient
Value: 2
Number of steps in each run: 1000
How many times the optimization must be run? : 10
First value of Lambda (parameter) <0.05> :
Maximum value of Lambda (parameter)<0.10> :
Select now SHAKE options
If you use SHAKE we suggest you to select
bond-stretchings only. Bond angle and
dihedrals are usually left free. OPTIONS:
NO SHAKE
Only the bonds which involve hydrogen atoms
will be constrained :
Constraints in IBH-JBH
All the bonds are constrained
(SHAKE) In addition IB-JB
Finally is is also possible to choose if the
bonds are constrained to the distance in the
first set of coordinates :
Length = CONSTR
Or by the distance defined by the TOPOLOGY :
Length = BO
Options
<1> SHAKE is not used
2 Constraints from IBH-JBH,length=BO
3 Constraints from IBH-JBH,length from CONSTR
4 In adition from IB-JB,length=BO
5 In adition from IB-JB,length from CONSTR
Value : 1
The calcul of the electric energy and
Van der Waals energy can be made by three
different methods :
By scanning, which means that each step
is calculated all the possible interactions
between atoms pairs inside a further especified
radius.
By interaction of previous selected atoms pairs
inside a further especified radius. In this
case it will be necessary to make a list of the
atoms inside such a distance, this list will
be made by the same program, but after several
steps it will change. The program will ask for
the number of steps to do it.
By interaction of previous selected groups pairs
inside a further especified radius. In this
case will be necessary to make a list of the
atoms inside such a distance, this list will
be made by the same program, but after several
steps it will change. The program will ask for
the number of steps to do it. And the groups are
already defined by the topology program.
Options
<1> By interaction between groups pair
2 By interaction between atoms pair
3 By scanning
4 By grid cells
Value : 1
After how many steps the list change <10> :
The program will ask now for the radius value
of cut-off & cut-on, RCUTL>RCUTP, as though
as the radius values for the switching function.
You can see :
Effects of Truncating Long Range Forces on
Protein Dynamics
Loncharich ,R.J. & Brooks ,B.R.
Division of Computer Research & Technology
National Institutes of Health (Bethesda)
PROTEINS (1989) 6 , pp 32-45
or
Cut-off size does strongly influence Molecular
Dynamics results on solvated polypeptides
Schreiber,H. & Steinhauser, O.
BIOCHEMISTRY (1991) 31 , pp 5856-5860
Value of RCUTP <0.8 nm> :
Value of RSWI2 <10.0 nm> :
Value of RCUI2 <10.0 nm> :
Value of RCUTL <1.3 nm> :
Sequence radius to calculate the interaction: 99999
At this point it will be possible to choose
the shape and also the size of the BOX.
Inside of this BOX the protein and molecules
surrounding will remain defined using the
BOUNDARY CONDITIONS algorithm .
Options
<1> No periodicity is taked into account
2 Octahedric BOX,BETA=90
3 Rombohedric BOX, chose BETA
Value : 1
At this point the program will ask for the different
constraints.
It will be necessary to especify the name of the file
which has the atoms to restrain. In that case these
must have been alocated in the working directory either
from the beginning or before you run the program.
The next questions will be related with the strength
of the constraints and the distance between atoms,
and to understand this questions read the
GROMOS manual before.
Options
<1> No position restraining
2 Position restraining using CHO
3 Idem using CHO/atomic B-factors
Value : 1
Options
<1> No distance restraining
2 Distance restraining using CDIS
3 Idem using CDIS*weight factors
Value : 1
Options
<1> No dihedral restraining
2 Restraining using CDLR*weight factors
Value : 1
Print energy every n-steps, n=: 100
Options:
<a> ====> Create INPUTS
b ====> RUN a process
c ====> ANALYSIS
d ====> NEW SIMULATION
e ====> Logout of this program
b
Options to RUN:
<a> ====> TOPOLOGY
b ====> ENERGY OPTIMIZATION
c ====> DYNAMIC
d ====> Go to MENU
b
Options:
<a> ====> Single System
b ====> Complex System
a
Have you run the TOPOLOGY program ? no/<yes> yes
Have you made the DYNAMIC input ? no/<yes> no
In this case you will have some errors, but the
the program will run perfectly well.
IF YOU ARE RUNNING A SYSTEM WITH WATER
AS SOLVENT YOU WILL HAVE PROBLEMS
BUILD THE DYNAMICS INPUT FIRST
Do you want continue with the optimization ? no/<yes> yes
Options :
<a> ====> Start the optimization
b ====> Continue the optimization
c ====> Optimize the water box
d ====> Optimize the counter-ions
e ====> Go to menu
a
Directory where is the work : /disc9/BE/e14910/practica_7/grumos-model2
Running a NIS simulation .
You will found the information about errors in file.err
cp: no se puede efectuar `stat' sobre `.grumosmodel2_md.dat_old': No such file or directory
cp: no se puede efectuar `stat' sobre `.grumosmodel2_md.cdat_old': No such file or directory
Options :
<a> ====> Create INPUTS
b ====> RUN a process
c ====> ANALYSIS
d ====> NEW SIMULATION
e ====> Logout of this program
e
|
Com hem pogut veure en el transcurs de l´execuxció del programa, els passos que hem seguit:
- Crear inputs per després córrer Topology.
- Córrer Topology amb les condicions: sistema simple, sense ponts S-S, sense solvent inert, amb cues als extrems (NH3 i COO-).
- Crear inputs per a després córrer Energy Optimitzation.
- Córrer Energy Optimitzation amb les condicions: sistema simple, steepest descent (descens cap al mínim de forma molt pronunciada), correrem el programa 10 cops en 1000 passos i n´imprimirem els valors cada 100 resultats, sense constricció dels àtoms (només contempla la constricció dels àtoms d´H), interacció entre grups de parells, sense periodicitat i sense optimització per diedres...
- RESULTATS OBTINGUTS DE L´APLICACIÓ DEL PROGRAMA ALS EXEMPLES COMENTATS I PROBLEMES PROPOSATS A LA PRÀCTICA
Els resultats obtinguts per al grumosmodel2.pdb, han estat els següents:
[e14910.bio.acexs.au.upf@au491 practica_7]$ cd grumos-model2
[e14910.bio.acexs.au.upf@au491 grumos-model2]$ ls
total 3048
4 coordinates/ 336 grumosmodel2_top.bin 20 outgrumosmodel2_emnum004.lis
4 energies/ 92 grumosmodel2.wvg 20 outgrumosmodel2_emnum005.lis
4 file.err 132 inicial.gsf 20 outgrumosmodel2_emnum006.lis
4 file.errpdb 24 outgrumosmodel2_emnum001.lis 20 outgrumosmodel2_emnum007.lis
132 grumosmodel2.hwvg 20 outgrumosmodel2_emnum002.lis 20 outgrumosmodel2_emnum008.lis
108 grumosmodel2.pdb 20 outgrumosmodel2_emnum003.lis 20 outgrumosmodel2_emnum009.lis
20 outgrumosmodel2_emnum010.lis 2024 outgrumosmodel2_top.lis
4 outgrumosmodel2_h.lis
[e14910.bio.acexs.au.upf@au491 grumos-model2]$
|
El que ens interessa observar d'aquests arxius de sortida, és si realment l'energia del sistema original ha disminuit (l'estructura s'ha relaxat i es troba en una conformació més estable).
Per a comprovar-ho cal observar el primer dels outputs generats (outgrumosmodel2_emnum001.lis) i l'últim (outgrumosmodel2_emnum010.lis), i comprovar si el valor de l'energia potencial total ha passat d'un valor positiu a un valor negatiu:
3. A T O M I C C O O R D I N A T E S :
grumosmodel2
Subrutina runem
SHAKE COORDINATES, NITER = 1
Subrutina Force
NUMBER OF NON-BONDED CHARGE GROUP PAIRS (I>J) = 49518
NJNB= 49518NTN= 2
STEP NIP1 NIS1 NIP2 NIS2 NIP3 NIS3 NITI STEP-SIZE [...]
E-POT-TOT E-BOND-H E-BOND E-ANGLE-H [...]
E-EL-G1-G1 E-EL-G1-G2 E-EL-G2-G2 E-EL-G1-G3 E-EL-G2-G3 E-EL-G3-G3 E-EL-G1-G4 [...]
E-LJ-G1-G1 E-LJ-G1-G2 E-LJ-G2-G2 E-LJ-G1-G3 E-LJ-G2-G3 E-LJ-G3-G3 E-LJ-G1-G4 [...]
E-DIS-R. E-POS-R. E-DIH-R.
0 1 0 0 0 0 0 0 0.5000E-01 [...]
0.13951E+05 0.1534E+02 0.1812E+04 0.4809E+03 [...]
-0.4697E+04 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 [...]
0.3951E+04 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 [...]
0.0000E+00 0.0000E+00 0.6815-269
[...]
STEP NIP1 NIS1 NIP2 NIS2 NIP3 NIS3 NITI STEP-SIZE [...]
E-POT-TOT E-BOND-H E-BOND E-ANGLE-H [...]
E-EL-G1-G1 E-EL-G1-G2 E-EL-G2-G2 E-EL-G1-G3 E-EL-G2-G3 E-EL-G3-G3 E-EL-G1-G4 [...]
E-LJ-G1-G1 E-LJ-G1-G2 E-LJ-G2-G2 E-LJ-G1-G3 E-LJ-G2-G3 E-LJ-G3-G3 E-LJ-G1-G4 [...]
E-DIS-R. E-POS-R. E-DIH-R.
1000 1 0 0 0 0 0 0 0.3273E-03 [...]
-0.15142E+05 0.1990E+02 0.1859E+03 0.1911E+03 [...]
-0.8314E+04 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 [...]
-0.9897E+04 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 0.0000E+00 [...]
0.0000E+00 0.0000E+00 0.6815-269
|
... i ara visualitzem els arxius continguts en el directori coordinates:
[e14910.bio.acexs.au.upf@au491 grumos-model2]$ cd coordinates/
[e14910.bio.acexs.au.upf@au491 coordinates]$ ls
total 5672
4 anglen.log 112 grumosmodel2xemnum006.gsf 160 modelopt2.out
4 bplot.log 112 grumosmodel2xemnum007.gsf 196 modelopt2.pdb
4 clean.log 112 grumosmodel2xemnum008.gsf 28 modelopt2.pln
108 grumosmodel2.pdb 112 grumosmodel2xemnum009.gsf 36 modelopt2.rin
112 grumosmodel2xemnum001.gsf 112 grumosmodel2xemnum010.gsf 4 modelopt2.sco
112 grumosmodel2xemnum002.gsf 112 grumosmodel2xvmd0000.gsf 28 modelopt2.sdh
112 grumosmodel2xemnum003.gsf 112 modelopt2.lan 32 nb.log
112 grumosmodel2xemnum004.gsf 76 modelopt2.nb 4 pplot.log
112 grumosmodel2xemnum005.gsf 180 modelopt2.new 4 secstr.log
4 tplot.log
[e14910.bio.acexs.au.upf@au491 coordinates]$
|
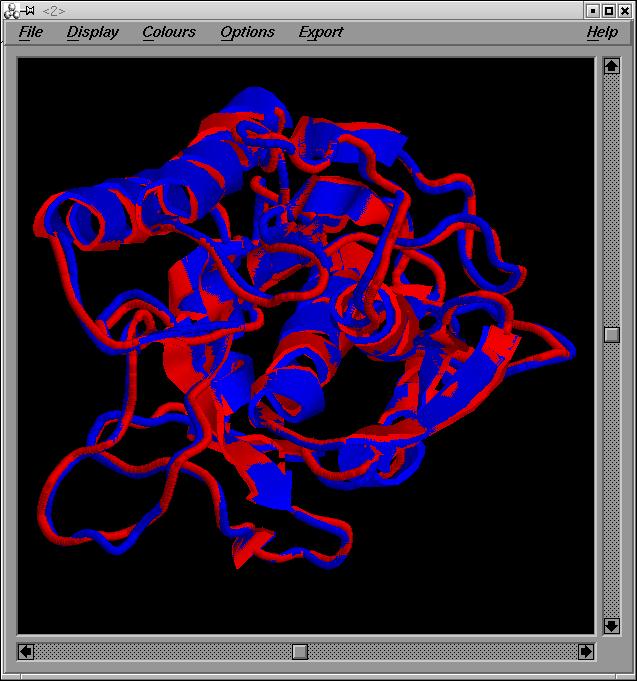
Per tal de comparar el meu model i el model optimitzat, correré el programa XAM de superposició estructural. Els arxius d'entrada per al programa són: grumosmodel2.pdb i grumosmodel2xvmd0010.gsf.
La sortida del programa genera els següents arxius de sortida:
- un valor de RMSD (xammodelsgrumos2)
- una estructura (xammodelsgrumos2.pdb)
Com es pot observar a partir de la imatge de les dues estructures superposades, ambdós models són força similars excepte en les regions dels loops: un cop més dir que aquestes regions són força variables.
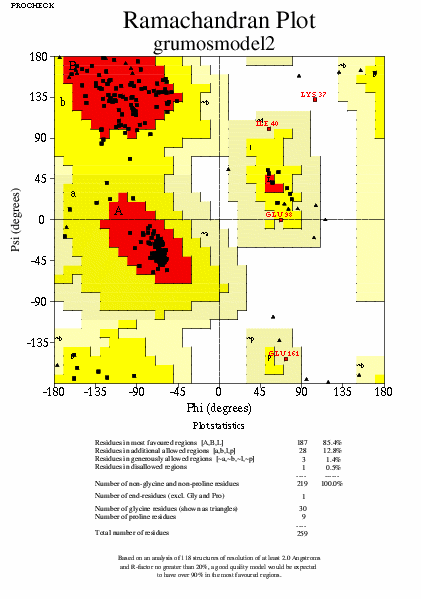
I finalment per a veure com de millor és el model optimitzat respecte l'original, correrem la comanda de procheck:
Tot i que en fer el procheck el model optimitzat hauria de sortir millor, en aquest cas no ha estat així. De totes maneres el model optimitzat té una energia menor que l'original (com a resultat de l'optimització) i per tant em de creure que és un model millorat.
|



{kind=link}