
Extending signalling pathways with protein-interaction networks. Application to apoptosis.
Joan Planas-Iglesias, Emre Guney, Javier Garcia-Garcia, Kevin A. Robertson, Sobia Raza, Tom C. Freeman, Peter Ghazal and Baldo Oliva.
OMICS. 2012 May;16(5):245-56. Epub 2012 Mar 2. (PMID: 22385281)
Here we present the supplementary materials and complete numerical results for the paper "Extending signalling pathways with protein-interaction networks. Application to apoptosis.". In this work, we build four different apoptotsis-related networks from the 53 hand-curated apoptotsis-related proteins known by 20051 (referred as seeds). These seeds are connected in networks according to the knowledge obtained from experimentally determined PPIs (Experimental network) or from these sources plus protein interaction predictions from STRING2 and a 3D-derived database3 (STRING+ network). On these two networks we perform interologs predictions4 obtaining the corresponding orthology expanded versions of the former networks (Experimental expansion network and STRING+ expansion network respectivelly). Briefly, a pair of human proteins are predicted to interact if there exist a described interaction between the correesponding orthologs in another organism. We annotate the interologs predictions with their Joint-eValue (JEV), the harmonic mean of the eValues between human proteins in the network and their corresponding interologs4.
On these networks we use different scoring methods to predict novel apoptosis proteins. In particular we use different topology-based scoring methods, content-of-information-based indicators, and the STRING combined score (SC)5. Among the topology-based scoring methods, we use Functional Flow (FF)6, Apoptosis Linkage Degree (ALD, as an analogous measure to Cancer Linkage Degree7 and iterative Apoptosis Linkage Degree (iALD). ALD is defined as the number of interactions with apoptosis proteins a given node in the network has. iALD mimics FF behaviour iteratively extending the scoring of ALD through the network (work in preparation). We obtain an initial list of candidates after performing a 5-fold cross-validation assay over the networks and selecting as candidates nodes that achieved 95% specificity and 90% PPV. Then, we compare our candidates with two independantly obtained validation lists of apoptosis involved proteins known during the period 2005-2010. We obtain these validation lists by KeyWord search using BIANA8 or using ProMiner text mining tool9.We obtain a p-Value for our predictions given by:
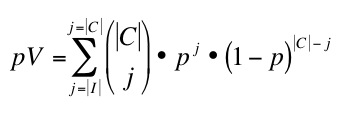
were P is the set of proteins in teh network, C the set of candiates, V the set of proteins in teh validation set and I the set of candidate proteins validated by text mining. The chance of success p corresponds to |V|/|P|. This p-Value is calculated for the candidates posed by each scoring system, the candidates posed by an increasing number of scoring methods (i.e. at least 2, or 3, or 4, etc.), and the candidates posed by all possible combinations of different scoring methods. We use Biana keyword search and FuncAssociate10 to annotate functions of candidates.
On the context of transferring functional annotation, we study the relevance of sequence similarity in the interologs prediction on ortholgy expanded networks. We adress this issue in two steps. First, we compare the differences between JEV distributions of the candidates and the rest of the network using a Kolmogorov-Smirnoff test. Then, we apply several orthology filters removing from the networks all predictions that do not achieve a certain JEV threshold and we compute the text mining validation p-Value of the remaining predictions. With this approach we can study whether these filters improve the predictions or not.